US20050002865A1 - Diagnostic/therapeutic agents - Google Patents
Diagnostic/therapeutic agents Download PDFInfo
- Publication number
- US20050002865A1 US20050002865A1 US10/734,730 US73473003A US2005002865A1 US 20050002865 A1 US20050002865 A1 US 20050002865A1 US 73473003 A US73473003 A US 73473003A US 2005002865 A1 US2005002865 A1 US 2005002865A1
- Authority
- US
- United States
- Prior art keywords
- agent
- vector
- acid
- gas
- microbubbles
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 239000003814 drug Substances 0.000 title claims description 46
- 229940124597 therapeutic agent Drugs 0.000 title description 6
- 229940039227 diagnostic agent Drugs 0.000 title description 5
- 239000000032 diagnostic agent Substances 0.000 title description 5
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 86
- 230000027455 binding Effects 0.000 claims abstract description 54
- 239000000463 material Substances 0.000 claims abstract description 27
- 239000007788 liquid Substances 0.000 claims abstract description 10
- 239000013543 active substance Substances 0.000 claims abstract description 9
- 239000000725 suspension Substances 0.000 claims abstract description 7
- 239000008365 aqueous carrier Substances 0.000 claims abstract description 4
- 239000013598 vector Substances 0.000 claims description 180
- 239000007789 gas Substances 0.000 claims description 57
- 108090000623 proteins and genes Proteins 0.000 claims description 45
- 102000004169 proteins and genes Human genes 0.000 claims description 42
- 210000004027 cell Anatomy 0.000 claims description 39
- 108091034117 Oligonucleotide Proteins 0.000 claims description 37
- 229940079593 drug Drugs 0.000 claims description 37
- 238000000034 method Methods 0.000 claims description 36
- 230000008685 targeting Effects 0.000 claims description 28
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 26
- 102000016611 Proteoglycans Human genes 0.000 claims description 25
- 108010067787 Proteoglycans Proteins 0.000 claims description 25
- 239000002872 contrast media Substances 0.000 claims description 25
- 239000000203 mixture Substances 0.000 claims description 24
- 230000001225 therapeutic effect Effects 0.000 claims description 22
- 150000001875 compounds Chemical class 0.000 claims description 21
- 230000003993 interaction Effects 0.000 claims description 20
- 239000003112 inhibitor Substances 0.000 claims description 19
- 150000003904 phospholipids Chemical class 0.000 claims description 18
- 230000008569 process Effects 0.000 claims description 18
- 239000012528 membrane Substances 0.000 claims description 16
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 15
- 239000004094 surface-active agent Substances 0.000 claims description 15
- 230000008878 coupling Effects 0.000 claims description 14
- 238000010168 coupling process Methods 0.000 claims description 14
- 238000005859 coupling reaction Methods 0.000 claims description 14
- 239000003102 growth factor Substances 0.000 claims description 14
- 102000004190 Enzymes Human genes 0.000 claims description 13
- 108090000790 Enzymes Proteins 0.000 claims description 13
- 238000003384 imaging method Methods 0.000 claims description 13
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 claims description 12
- 230000000975 bioactive effect Effects 0.000 claims description 12
- 230000015572 biosynthetic process Effects 0.000 claims description 12
- 238000002604 ultrasonography Methods 0.000 claims description 11
- 210000002889 endothelial cell Anatomy 0.000 claims description 10
- 150000002148 esters Chemical class 0.000 claims description 10
- 239000000126 substance Substances 0.000 claims description 10
- 229960002685 biotin Drugs 0.000 claims description 9
- 239000011616 biotin Substances 0.000 claims description 9
- 238000009472 formulation Methods 0.000 claims description 9
- 108091005804 Peptidases Proteins 0.000 claims description 8
- 229950003499 fibrin Drugs 0.000 claims description 8
- 108010067225 Cell Adhesion Molecules Proteins 0.000 claims description 7
- 102000009123 Fibrin Human genes 0.000 claims description 7
- 108010073385 Fibrin Proteins 0.000 claims description 7
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 claims description 7
- 239000004365 Protease Substances 0.000 claims description 7
- 230000021164 cell adhesion Effects 0.000 claims description 7
- 238000001727 in vivo Methods 0.000 claims description 7
- 150000002576 ketones Chemical class 0.000 claims description 7
- 239000003446 ligand Substances 0.000 claims description 7
- 238000002360 preparation method Methods 0.000 claims description 7
- 102000016289 Cell Adhesion Molecules Human genes 0.000 claims description 6
- 239000000813 peptide hormone Substances 0.000 claims description 6
- 102000004127 Cytokines Human genes 0.000 claims description 5
- 108090000695 Cytokines Proteins 0.000 claims description 5
- 230000004568 DNA-binding Effects 0.000 claims description 5
- 108010000499 Thromboplastin Proteins 0.000 claims description 5
- 102000002262 Thromboplastin Human genes 0.000 claims description 5
- 238000005345 coagulation Methods 0.000 claims description 5
- 230000015271 coagulation Effects 0.000 claims description 5
- 229940088597 hormone Drugs 0.000 claims description 5
- 239000005556 hormone Substances 0.000 claims description 5
- 238000000338 in vitro Methods 0.000 claims description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 4
- 239000004215 Carbon black (E152) Substances 0.000 claims description 4
- 229930195733 hydrocarbon Natural products 0.000 claims description 4
- 150000002430 hydrocarbons Chemical class 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 230000002209 hydrophobic effect Effects 0.000 claims description 4
- KAVGMUDTWQVPDF-UHFFFAOYSA-N perflubutane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)F KAVGMUDTWQVPDF-UHFFFAOYSA-N 0.000 claims description 4
- 150000008106 phosphatidylserines Chemical class 0.000 claims description 4
- 239000000758 substrate Substances 0.000 claims description 4
- 230000003444 anaesthetic effect Effects 0.000 claims description 3
- 230000001772 anti-angiogenic effect Effects 0.000 claims description 3
- 239000002246 antineoplastic agent Substances 0.000 claims description 3
- 239000011230 binding agent Substances 0.000 claims description 3
- 230000008512 biological response Effects 0.000 claims description 3
- 230000001413 cellular effect Effects 0.000 claims description 3
- 239000003638 chemical reducing agent Substances 0.000 claims description 3
- 150000002321 glycerophosphoglycerophosphoglycerols Chemical class 0.000 claims description 3
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 3
- 239000011261 inert gas Substances 0.000 claims description 3
- QYSGYZVSCZSLHT-UHFFFAOYSA-N octafluoropropane Chemical compound FC(F)(F)C(F)(F)C(F)(F)F QYSGYZVSCZSLHT-UHFFFAOYSA-N 0.000 claims description 3
- NJCBUSHGCBERSK-UHFFFAOYSA-N perfluoropentane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F NJCBUSHGCBERSK-UHFFFAOYSA-N 0.000 claims description 3
- 150000008103 phosphatidic acids Chemical class 0.000 claims description 3
- 229940067626 phosphatidylinositols Drugs 0.000 claims description 3
- 150000003905 phosphatidylinositols Chemical class 0.000 claims description 3
- 230000002285 radioactive effect Effects 0.000 claims description 3
- SFZCNBIFKDRMGX-UHFFFAOYSA-N sulfur hexafluoride Chemical compound FS(F)(F)(F)(F)F SFZCNBIFKDRMGX-UHFFFAOYSA-N 0.000 claims description 3
- 229960000909 sulfur hexafluoride Drugs 0.000 claims description 3
- 230000003110 anti-inflammatory effect Effects 0.000 claims description 2
- 230000000884 anti-protozoa Effects 0.000 claims description 2
- 230000003356 anti-rheumatic effect Effects 0.000 claims description 2
- 230000002365 anti-tubercular Effects 0.000 claims description 2
- 239000000043 antiallergic agent Substances 0.000 claims description 2
- 229940121375 antifungal agent Drugs 0.000 claims description 2
- 239000003429 antifungal agent Substances 0.000 claims description 2
- 229940034982 antineoplastic agent Drugs 0.000 claims description 2
- 239000003435 antirheumatic agent Substances 0.000 claims description 2
- 239000010836 blood and blood product Substances 0.000 claims description 2
- 229940125691 blood product Drugs 0.000 claims description 2
- 229940097217 cardiac glycoside Drugs 0.000 claims description 2
- 239000002368 cardiac glycoside Substances 0.000 claims description 2
- 238000004581 coalescence Methods 0.000 claims description 2
- 150000002170 ethers Chemical class 0.000 claims description 2
- 230000005291 magnetic effect Effects 0.000 claims description 2
- 230000002503 metabolic effect Effects 0.000 claims description 2
- 239000000842 neuromuscular blocking agent Substances 0.000 claims description 2
- 229910052757 nitrogen Inorganic materials 0.000 claims description 2
- 229940127240 opiate Drugs 0.000 claims description 2
- 229960004065 perflutren Drugs 0.000 claims description 2
- 239000002861 polymer material Substances 0.000 claims description 2
- 239000000523 sample Substances 0.000 claims description 2
- 239000000932 sedative agent Substances 0.000 claims description 2
- 229930002534 steroid glycoside Natural products 0.000 claims description 2
- QTJXVIKNLHZIKL-UHFFFAOYSA-N sulfur difluoride Chemical compound FSF QTJXVIKNLHZIKL-UHFFFAOYSA-N 0.000 claims description 2
- TXEYQDLBPFQVAA-UHFFFAOYSA-N tetrafluoromethane Chemical compound FC(F)(F)F TXEYQDLBPFQVAA-UHFFFAOYSA-N 0.000 claims description 2
- 230000004797 therapeutic response Effects 0.000 claims description 2
- 229940088594 vitamin Drugs 0.000 claims description 2
- 229930003231 vitamin Natural products 0.000 claims description 2
- 235000013343 vitamin Nutrition 0.000 claims description 2
- 239000011782 vitamin Substances 0.000 claims description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 claims 1
- 239000003570 air Substances 0.000 claims 1
- 230000000840 anti-viral effect Effects 0.000 claims 1
- 230000003115 biocidal effect Effects 0.000 claims 1
- UBAZGMLMVVQSCD-UHFFFAOYSA-N carbon dioxide;molecular oxygen Chemical compound O=O.O=C=O UBAZGMLMVVQSCD-UHFFFAOYSA-N 0.000 claims 1
- 102000008395 cell adhesion mediator activity proteins Human genes 0.000 claims 1
- 230000020764 fibrinolysis Effects 0.000 claims 1
- 238000011835 investigation Methods 0.000 claims 1
- 239000003607 modifier Substances 0.000 claims 1
- 230000003533 narcotic effect Effects 0.000 claims 1
- LPMXVESGRSUGHW-HBYQJFLCSA-N ouabain Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@@H]1C[C@@]2(O)CC[C@H]3[C@@]4(O)CC[C@H](C=5COC(=O)C=5)[C@@]4(C)C[C@@H](O)[C@@H]3[C@@]2(CO)[C@H](O)C1 LPMXVESGRSUGHW-HBYQJFLCSA-N 0.000 claims 1
- 229960004692 perflenapent Drugs 0.000 claims 1
- 229950003332 perflubutane Drugs 0.000 claims 1
- 230000001624 sedative effect Effects 0.000 claims 1
- 229910052711 selenium Inorganic materials 0.000 claims 1
- 239000011669 selenium Substances 0.000 claims 1
- 229940124549 vasodilator Drugs 0.000 claims 1
- 239000003071 vasodilator agent Substances 0.000 claims 1
- 150000003722 vitamin derivatives Chemical class 0.000 claims 1
- 239000002961 echo contrast media Substances 0.000 abstract description 14
- 206010028980 Neoplasm Diseases 0.000 description 158
- -1 dipalmitoyllysine Chemical class 0.000 description 126
- 206010061218 Inflammation Diseases 0.000 description 81
- 230000004054 inflammatory process Effects 0.000 description 81
- 102000005962 receptors Human genes 0.000 description 62
- 108020003175 receptors Proteins 0.000 description 62
- 235000018102 proteins Nutrition 0.000 description 35
- 125000005647 linker group Chemical group 0.000 description 27
- 210000003038 endothelium Anatomy 0.000 description 19
- 102000006495 integrins Human genes 0.000 description 19
- 108010044426 integrins Proteins 0.000 description 19
- 238000006243 chemical reaction Methods 0.000 description 18
- 125000003396 thiol group Chemical group [H]S* 0.000 description 18
- 210000001519 tissue Anatomy 0.000 description 17
- 210000004165 myocardium Anatomy 0.000 description 16
- 125000006850 spacer group Chemical group 0.000 description 16
- 230000033115 angiogenesis Effects 0.000 description 15
- 239000003153 chemical reaction reagent Substances 0.000 description 15
- 230000012010 growth Effects 0.000 description 15
- 210000004379 membrane Anatomy 0.000 description 14
- 239000004005 microsphere Substances 0.000 description 14
- 229920001223 polyethylene glycol Polymers 0.000 description 14
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 13
- 108020004414 DNA Proteins 0.000 description 13
- 201000010099 disease Diseases 0.000 description 12
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 12
- 229940088598 enzyme Drugs 0.000 description 12
- 239000002253 acid Substances 0.000 description 11
- 125000003277 amino group Chemical group 0.000 description 11
- 239000000427 antigen Substances 0.000 description 11
- 108091007433 antigens Proteins 0.000 description 11
- 102000036639 antigens Human genes 0.000 description 11
- 235000014633 carbohydrates Nutrition 0.000 description 11
- 210000002865 immune cell Anatomy 0.000 description 11
- 230000001965 increasing effect Effects 0.000 description 11
- 229920000642 polymer Polymers 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 125000000524 functional group Chemical group 0.000 description 10
- 150000002632 lipids Chemical class 0.000 description 10
- 208000019553 vascular disease Diseases 0.000 description 10
- 201000011510 cancer Diseases 0.000 description 9
- 210000003734 kidney Anatomy 0.000 description 9
- 239000002502 liposome Substances 0.000 description 9
- 108010028921 Lipopeptides Proteins 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 8
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 7
- 206010028851 Necrosis Diseases 0.000 description 7
- 102000035195 Peptidases Human genes 0.000 description 7
- 239000005557 antagonist Substances 0.000 description 7
- 235000020958 biotin Nutrition 0.000 description 7
- 210000004556 brain Anatomy 0.000 description 7
- 229920001577 copolymer Polymers 0.000 description 7
- 230000002107 myocardial effect Effects 0.000 description 7
- 230000017074 necrotic cell death Effects 0.000 description 7
- 230000009257 reactivity Effects 0.000 description 7
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 6
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 150000001720 carbohydrates Chemical class 0.000 description 6
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 6
- 239000012634 fragment Substances 0.000 description 6
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 6
- 229940127126 plasminogen activator Drugs 0.000 description 6
- 210000004881 tumor cell Anatomy 0.000 description 6
- IYMAXBFPHPZYIK-BQBZGAKWSA-N Arg-Gly-Asp Chemical compound NC(N)=NCCC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(O)=O IYMAXBFPHPZYIK-BQBZGAKWSA-N 0.000 description 5
- 108090001008 Avidin Proteins 0.000 description 5
- 102000004225 Cathepsin B Human genes 0.000 description 5
- 108090000712 Cathepsin B Proteins 0.000 description 5
- 108010035532 Collagen Proteins 0.000 description 5
- 102000008186 Collagen Human genes 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 102100037362 Fibronectin Human genes 0.000 description 5
- 108010067306 Fibronectins Proteins 0.000 description 5
- 108010006035 Metalloproteases Proteins 0.000 description 5
- 102000005741 Metalloproteases Human genes 0.000 description 5
- 108010001014 Plasminogen Activators Proteins 0.000 description 5
- 102000001938 Plasminogen Activators Human genes 0.000 description 5
- 238000010976 amide bond formation reaction Methods 0.000 description 5
- 210000004204 blood vessel Anatomy 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 150000001718 carbodiimides Chemical class 0.000 description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 5
- 229920001436 collagen Polymers 0.000 description 5
- 239000000306 component Substances 0.000 description 5
- 238000012377 drug delivery Methods 0.000 description 5
- 210000004185 liver Anatomy 0.000 description 5
- 210000001165 lymph node Anatomy 0.000 description 5
- 210000000865 mononuclear phagocyte system Anatomy 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 229920001282 polysaccharide Polymers 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 230000002792 vascular Effects 0.000 description 5
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 4
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 4
- 102100032912 CD44 antigen Human genes 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- 102000014914 Carrier Proteins Human genes 0.000 description 4
- 229920002307 Dextran Polymers 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 4
- 229920002971 Heparan sulfate Polymers 0.000 description 4
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 4
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 4
- 108010042918 Integrin alpha5beta1 Proteins 0.000 description 4
- 108010085895 Laminin Proteins 0.000 description 4
- 102000007547 Laminin Human genes 0.000 description 4
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 4
- AUNGANRZJHBGPY-SCRDCRAPSA-N Riboflavin Chemical compound OC[C@@H](O)[C@@H](O)[C@@H](O)CN1C=2C=C(C)C(C)=CC=2N=C2C1=NC(=O)NC2=O AUNGANRZJHBGPY-SCRDCRAPSA-N 0.000 description 4
- 102000002938 Thrombospondin Human genes 0.000 description 4
- 108060008245 Thrombospondin Proteins 0.000 description 4
- 108010031318 Vitronectin Proteins 0.000 description 4
- 102100035140 Vitronectin Human genes 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 150000001350 alkyl halides Chemical class 0.000 description 4
- 150000001413 amino acids Chemical group 0.000 description 4
- VIROVYVQCGLCII-UHFFFAOYSA-N amobarbital Chemical compound CC(C)CCC1(CC)C(=O)NC(=O)NC1=O VIROVYVQCGLCII-UHFFFAOYSA-N 0.000 description 4
- 230000001656 angiogenetic effect Effects 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 229920001400 block copolymer Polymers 0.000 description 4
- 230000008499 blood brain barrier function Effects 0.000 description 4
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 4
- 229960004630 chlorambucil Drugs 0.000 description 4
- 235000012000 cholesterol Nutrition 0.000 description 4
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- AEUTYOVWOVBAKS-UWVGGRQHSA-N ethambutol Chemical compound CC[C@@H](CO)NCCN[C@@H](CC)CO AEUTYOVWOVBAKS-UWVGGRQHSA-N 0.000 description 4
- 210000002950 fibroblast Anatomy 0.000 description 4
- 239000007850 fluorescent dye Substances 0.000 description 4
- 235000019152 folic acid Nutrition 0.000 description 4
- 239000011724 folic acid Substances 0.000 description 4
- 150000004676 glycans Chemical class 0.000 description 4
- 229920000669 heparin Polymers 0.000 description 4
- 229960002897 heparin Drugs 0.000 description 4
- 239000000787 lecithin Substances 0.000 description 4
- 235000010445 lecithin Nutrition 0.000 description 4
- 210000002540 macrophage Anatomy 0.000 description 4
- 239000011859 microparticle Substances 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- 239000005017 polysaccharide Substances 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 238000006722 reduction reaction Methods 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 4
- UCTWMZQNUQWSLP-VIFPVBQESA-N (R)-adrenaline Chemical compound CNC[C@H](O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-VIFPVBQESA-N 0.000 description 3
- KIUMMUBSPKGMOY-UHFFFAOYSA-N 3,3'-Dithiobis(6-nitrobenzoic acid) Chemical compound C1=C([N+]([O-])=O)C(C(=O)O)=CC(SSC=2C=C(C(=CC=2)[N+]([O-])=O)C(O)=O)=C1 KIUMMUBSPKGMOY-UHFFFAOYSA-N 0.000 description 3
- 102000009027 Albumins Human genes 0.000 description 3
- 108010088751 Albumins Proteins 0.000 description 3
- 108010006654 Bleomycin Proteins 0.000 description 3
- 208000020084 Bone disease Diseases 0.000 description 3
- 208000003174 Brain Neoplasms Diseases 0.000 description 3
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 3
- 101100289995 Caenorhabditis elegans mac-1 gene Proteins 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- 229920000858 Cyclodextrin Polymers 0.000 description 3
- 229920000045 Dermatan sulfate Polymers 0.000 description 3
- WDJUZGPOPHTGOT-OAXVISGBSA-N Digitoxin Natural products O([C@H]1[C@@H](C)O[C@@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@@](C)([C@H](C6=CC(=O)OC6)CC5)CC4)CC3)CC2)C[C@H]1O)[C@H]1O[C@@H](C)[C@H](O[C@H]2O[C@@H](C)[C@@H](O)[C@@H](O)C2)[C@@H](O)C1 WDJUZGPOPHTGOT-OAXVISGBSA-N 0.000 description 3
- 102000002045 Endothelin Human genes 0.000 description 3
- 108050009340 Endothelin Proteins 0.000 description 3
- 108010024636 Glutathione Proteins 0.000 description 3
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 3
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 3
- 102100025323 Integrin alpha-1 Human genes 0.000 description 3
- 102100032817 Integrin alpha-5 Human genes 0.000 description 3
- 102100032816 Integrin alpha-6 Human genes 0.000 description 3
- 102100022339 Integrin alpha-L Human genes 0.000 description 3
- 108010041341 Integrin alpha1 Proteins 0.000 description 3
- 108010055795 Integrin alpha1beta1 Proteins 0.000 description 3
- 108010017642 Integrin alpha2beta1 Proteins 0.000 description 3
- 108010072255 Integrin alpha3beta1 Proteins 0.000 description 3
- 108010008212 Integrin alpha4beta1 Proteins 0.000 description 3
- 108010041014 Integrin alpha5 Proteins 0.000 description 3
- 108010041100 Integrin alpha6 Proteins 0.000 description 3
- 108010030465 Integrin alpha6beta1 Proteins 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 108010001831 LDL receptors Proteins 0.000 description 3
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 3
- 108010064548 Lymphocyte Function-Associated Antigen-1 Proteins 0.000 description 3
- 101150044441 PECAM1 gene Proteins 0.000 description 3
- 229930012538 Paclitaxel Natural products 0.000 description 3
- 102000013566 Plasminogen Human genes 0.000 description 3
- 108010051456 Plasminogen Proteins 0.000 description 3
- 108010039918 Polylysine Proteins 0.000 description 3
- 108090000184 Selectins Proteins 0.000 description 3
- 102000003800 Selectins Human genes 0.000 description 3
- 108010056088 Somatostatin Proteins 0.000 description 3
- 102000005157 Somatostatin Human genes 0.000 description 3
- 108010023197 Streptokinase Proteins 0.000 description 3
- 102000007000 Tenascin Human genes 0.000 description 3
- 108010008125 Tenascin Proteins 0.000 description 3
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 3
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 3
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 3
- 102100039037 Vascular endothelial growth factor A Human genes 0.000 description 3
- VRGWBRLULZUWAJ-XFFXIZSCSA-N [(2s)-2-[(1r,3z,5s,8z,12z,15s)-5,17-dihydroxy-4,8,12,15-tetramethyl-16-oxo-18-bicyclo[13.3.0]octadeca-3,8,12,17-tetraenyl]propyl] acetate Chemical compound C1\C=C(C)/CC\C=C(C)/CC[C@H](O)\C(C)=C/C[C@@H]2C([C@@H](COC(C)=O)C)=C(O)C(=O)[C@]21C VRGWBRLULZUWAJ-XFFXIZSCSA-N 0.000 description 3
- 229940022663 acetate Drugs 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 229960005305 adenosine Drugs 0.000 description 3
- AVJBPWGFOQAPRH-FWMKGIEWSA-N alpha-L-IdopA-(1->3)-beta-D-GalpNAc4S Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@H](OS(O)(=O)=O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](C(O)=O)O1 AVJBPWGFOQAPRH-FWMKGIEWSA-N 0.000 description 3
- 108010072041 arginyl-glycyl-aspartic acid Proteins 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000001580 bacterial effect Effects 0.000 description 3
- 210000002469 basement membrane Anatomy 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 229960001561 bleomycin Drugs 0.000 description 3
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 3
- 229960005069 calcium Drugs 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical class C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 230000000295 complement effect Effects 0.000 description 3
- 230000021615 conjugation Effects 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- RMRCNWBMXRMIRW-BYFNXCQMSA-M cyanocobalamin Chemical compound N#C[Co+]N([C@]1([H])[C@H](CC(N)=O)[C@]\2(CCC(=O)NC[C@H](C)OP(O)(=O)OC3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)C)C/2=C(C)\C([C@H](C/2(C)C)CCC(N)=O)=N\C\2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O RMRCNWBMXRMIRW-BYFNXCQMSA-M 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000001212 derivatisation Methods 0.000 description 3
- 229940051593 dermatan sulfate Drugs 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 3
- 229960003957 dexamethasone Drugs 0.000 description 3
- RMEDXOLNCUSCGS-UHFFFAOYSA-N droperidol Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CC=C(N2C(NC3=CC=CC=C32)=O)CC1 RMEDXOLNCUSCGS-UHFFFAOYSA-N 0.000 description 3
- 229960000394 droperidol Drugs 0.000 description 3
- 230000003511 endothelial effect Effects 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- 238000001215 fluorescent labelling Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- VRGWBRLULZUWAJ-UHFFFAOYSA-N fusaproliferin Natural products C1C=C(C)CCC=C(C)CCC(O)C(C)=CCC2C(C(COC(C)=O)C)=C(O)C(=O)C21C VRGWBRLULZUWAJ-UHFFFAOYSA-N 0.000 description 3
- 229920002674 hyaluronan Polymers 0.000 description 3
- 210000000987 immune system Anatomy 0.000 description 3
- 230000002608 insulinlike Effects 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- 108020004707 nucleic acids Proteins 0.000 description 3
- 102000039446 nucleic acids Human genes 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 125000000962 organic group Chemical group 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 229960001592 paclitaxel Drugs 0.000 description 3
- 230000007170 pathology Effects 0.000 description 3
- 229920000656 polylysine Polymers 0.000 description 3
- 229920002635 polyurethane Polymers 0.000 description 3
- 239000004814 polyurethane Substances 0.000 description 3
- 229930185346 proliferin Natural products 0.000 description 3
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 3
- 235000019419 proteases Nutrition 0.000 description 3
- 230000009145 protein modification Effects 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 3
- 229960000553 somatostatin Drugs 0.000 description 3
- 210000000952 spleen Anatomy 0.000 description 3
- 229960005202 streptokinase Drugs 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 230000001131 transforming effect Effects 0.000 description 3
- 238000012285 ultrasound imaging Methods 0.000 description 3
- 210000005166 vasculature Anatomy 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- 229910001868 water Inorganic materials 0.000 description 3
- KWGRBVOPPLSCSI-WPRPVWTQSA-N (-)-ephedrine Chemical compound CN[C@@H](C)[C@H](O)C1=CC=CC=C1 KWGRBVOPPLSCSI-WPRPVWTQSA-N 0.000 description 2
- NGGMYCMLYOUNGM-UHFFFAOYSA-N (-)-fumagillin Natural products O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)C=CC=CC=CC=CC(O)=O)CCC21CO2 NGGMYCMLYOUNGM-UHFFFAOYSA-N 0.000 description 2
- PROQIPRRNZUXQM-UHFFFAOYSA-N (16alpha,17betaOH)-Estra-1,3,5(10)-triene-3,16,17-triol Natural products OC1=CC=C2C3CCC(C)(C(C(O)C4)O)C4C3CCC2=C1 PROQIPRRNZUXQM-UHFFFAOYSA-N 0.000 description 2
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 2
- GDTZPEHTYWXYAR-XGXHKTLJSA-N (8r,9s,10r,13s,14s,17r)-17-acetyl-17-hydroxy-13-methyl-1,2,8,9,10,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-3-one Chemical compound C1=CC2=CC(=O)CC[C@@H]2[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 GDTZPEHTYWXYAR-XGXHKTLJSA-N 0.000 description 2
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 2
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 description 2
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 2
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- ZEHYJZXQEQOSON-AATRIKPKSA-N (e)-1-chloro-3-ethylpent-1-en-4-yn-3-ol Chemical compound CCC(O)(C#C)\C=C\Cl ZEHYJZXQEQOSON-AATRIKPKSA-N 0.000 description 2
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 2
- JIHQDMXYYFUGFV-UHFFFAOYSA-N 1,3,5-triazine Chemical class C1=NC=NC=N1 JIHQDMXYYFUGFV-UHFFFAOYSA-N 0.000 description 2
- MEZJQXVOMGUAMP-UHFFFAOYSA-N 1-(2-methylnaphthalen-1-yl)pyrrole-2,5-dione Chemical class CC1=CC=C2C=CC=CC2=C1N1C(=O)C=CC1=O MEZJQXVOMGUAMP-UHFFFAOYSA-N 0.000 description 2
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 2
- LEBVLXFERQHONN-UHFFFAOYSA-N 1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide Chemical compound CCCCN1CCCCC1C(=O)NC1=C(C)C=CC=C1C LEBVLXFERQHONN-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 2
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 2
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 2
- ZEEKNRMEDJKDMO-UHFFFAOYSA-N 2-cyclohexyl-3,5-dimethylphenol Chemical compound OC1=CC(C)=CC(C)=C1C1CCCCC1 ZEEKNRMEDJKDMO-UHFFFAOYSA-N 0.000 description 2
- SQVIAVUSQAWMKL-UHFFFAOYSA-N 3-[2-(ethylamino)-1-hydroxyethyl]phenol Chemical compound CCNCC(O)C1=CC=CC(O)=C1 SQVIAVUSQAWMKL-UHFFFAOYSA-N 0.000 description 2
- CFKMVGJGLGKFKI-UHFFFAOYSA-N 4-chloro-m-cresol Chemical compound CC1=CC(O)=CC=C1Cl CFKMVGJGLGKFKI-UHFFFAOYSA-N 0.000 description 2
- NTNZDKLXIBVSQQ-UHFFFAOYSA-N 4-methyl-7-prop-2-ynoxychromen-2-one Chemical compound C1=C(OCC#C)C=CC2=C1OC(=O)C=C2C NTNZDKLXIBVSQQ-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- FTOAOBMCPZCFFF-UHFFFAOYSA-N 5,5-diethylbarbituric acid Chemical compound CCC1(CC)C(=O)NC(=O)NC1=O FTOAOBMCPZCFFF-UHFFFAOYSA-N 0.000 description 2
- APKFDSVGJQXUKY-KKGHZKTASA-N Amphotericin-B Natural products O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1C=CC=CC=CC=CC=CC=CC=C[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-KKGHZKTASA-N 0.000 description 2
- 102400000068 Angiostatin Human genes 0.000 description 2
- 108010079709 Angiostatins Proteins 0.000 description 2
- 102400000345 Angiotensin-2 Human genes 0.000 description 2
- 101800000733 Angiotensin-2 Proteins 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- 102000000412 Annexin Human genes 0.000 description 2
- 108050008874 Annexin Proteins 0.000 description 2
- 108090000672 Annexin A5 Proteins 0.000 description 2
- 102000004121 Annexin A5 Human genes 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 108010024976 Asparaginase Proteins 0.000 description 2
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 2
- KUVIULQEHSCUHY-XYWKZLDCSA-N Beclometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O KUVIULQEHSCUHY-XYWKZLDCSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- QORQZMBCPRBCAB-UHFFFAOYSA-M Butabarbital sodium Chemical compound [Na+].CCC(C)C1(CC)C(=O)NC([O-])=NC1=O QORQZMBCPRBCAB-UHFFFAOYSA-M 0.000 description 2
- QFOHBWFCKVYLES-UHFFFAOYSA-N Butylparaben Chemical compound CCCCOC(=O)C1=CC=C(O)C=C1 QFOHBWFCKVYLES-UHFFFAOYSA-N 0.000 description 2
- AUJXLBOHYWTPFV-BLWRDSOESA-N CS[C@H]1SC[C@H]2N(C)C(=O)[C@@H](C)NC(=O)[C@H](COC(=O)[C@@H](C(C)C)N(C)C(=O)[C@@H]1N(C)C(=O)[C@@H](C)NC(=O)[C@H](COC(=O)[C@@H](C(C)C)N(C)C2=O)NC(=O)c1cnc2ccccc2n1)NC(=O)c1cnc2ccccc2n1 Chemical compound CS[C@H]1SC[C@H]2N(C)C(=O)[C@@H](C)NC(=O)[C@H](COC(=O)[C@@H](C(C)C)N(C)C(=O)[C@@H]1N(C)C(=O)[C@@H](C)NC(=O)[C@H](COC(=O)[C@@H](C(C)C)N(C)C2=O)NC(=O)c1cnc2ccccc2n1)NC(=O)c1cnc2ccccc2n1 AUJXLBOHYWTPFV-BLWRDSOESA-N 0.000 description 2
- 102400000113 Calcitonin Human genes 0.000 description 2
- 108060001064 Calcitonin Proteins 0.000 description 2
- 108010065839 Capreomycin Proteins 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 102000019034 Chemokines Human genes 0.000 description 2
- 108010012236 Chemokines Proteins 0.000 description 2
- 229920001661 Chitosan Polymers 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- ITRJWOMZKQRYTA-RFZYENFJSA-N Cortisone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)CC2=O ITRJWOMZKQRYTA-RFZYENFJSA-N 0.000 description 2
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- AUNGANRZJHBGPY-UHFFFAOYSA-N D-Lyxoflavin Natural products OCC(O)C(O)C(O)CN1C=2C=C(C)C(C)=CC=2N=C2C1=NC(=O)NC2=O AUNGANRZJHBGPY-UHFFFAOYSA-N 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 2
- 240000001879 Digitalis lutea Species 0.000 description 2
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 2
- IIUZTXTZRGLYTI-UHFFFAOYSA-N Dihydrogriseofulvin Natural products COC1CC(=O)CC(C)C11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 IIUZTXTZRGLYTI-UHFFFAOYSA-N 0.000 description 2
- ASXBYYWOLISCLQ-UHFFFAOYSA-N Dihydrostreptomycin Natural products O1C(CO)C(O)C(O)C(NC)C1OC1C(CO)(O)C(C)OC1OC1C(N=C(N)N)C(O)C(N=C(N)N)C(O)C1O ASXBYYWOLISCLQ-UHFFFAOYSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- 108010024212 E-Selectin Proteins 0.000 description 2
- 102100023471 E-selectin Human genes 0.000 description 2
- 108010009858 Echinomycin Proteins 0.000 description 2
- 108010061435 Enalapril Proteins 0.000 description 2
- VTUSIVBDOCDNHS-UHFFFAOYSA-N Etidocaine Chemical compound CCCN(CC)C(CC)C(=O)NC1=C(C)C=CC=C1C VTUSIVBDOCDNHS-UHFFFAOYSA-N 0.000 description 2
- 108010049003 Fibrinogen Proteins 0.000 description 2
- 102000008946 Fibrinogen Human genes 0.000 description 2
- ZIIJJOPLRSCQNX-UHFFFAOYSA-N Flurazepam hydrochloride Chemical compound Cl.Cl.N=1CC(=O)N(CCN(CC)CC)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F ZIIJJOPLRSCQNX-UHFFFAOYSA-N 0.000 description 2
- UGJMXCAKCUNAIE-UHFFFAOYSA-N Gabapentin Chemical compound OC(=O)CC1(CN)CCCCC1 UGJMXCAKCUNAIE-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- XURCMZMFZXXQDJ-UKNJCJGYSA-N Gestonorone caproate Chemical compound C1CC2=CC(=O)CC[C@@H]2[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)CCCCC)[C@@]1(C)CC2 XURCMZMFZXXQDJ-UKNJCJGYSA-N 0.000 description 2
- BJJXHLWLUDYTGC-ANULTFPQSA-N Gestrinone Chemical compound C1CC(=O)C=C2CC[C@@H]([C@H]3[C@@](CC)([C@](CC3)(O)C#C)C=C3)C3=C21 BJJXHLWLUDYTGC-ANULTFPQSA-N 0.000 description 2
- GZZJHPZDXZCDDA-UHFFFAOYSA-N Gitaloxin Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CC(C(C6(C)CC5)C=5COC(=O)C=5)OC=O)O)CC4)(C)CC3)CC2O)C)CC1O GZZJHPZDXZCDDA-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- JMBQKKAJIKAWKF-UHFFFAOYSA-N Glutethimide Chemical compound C=1C=CC=CC=1C1(CC)CCC(=O)NC1=O JMBQKKAJIKAWKF-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 244000068988 Glycine max Species 0.000 description 2
- 235000010469 Glycine max Nutrition 0.000 description 2
- 229920002527 Glycogen Polymers 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- UXWOXTQWVMFRSE-UHFFFAOYSA-N Griseoviridin Natural products O=C1OC(C)CC=C(C(NCC=CC=CC(O)CC(O)C2)=O)SCC1NC(=O)C1=COC2=N1 UXWOXTQWVMFRSE-UHFFFAOYSA-N 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 108091006905 Human Serum Albumin Proteins 0.000 description 2
- 102000008100 Human Serum Albumin Human genes 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 102000006992 Interferon-alpha Human genes 0.000 description 2
- 108010047761 Interferon-alpha Proteins 0.000 description 2
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 102100026262 Metalloproteinase inhibitor 2 Human genes 0.000 description 2
- NRFJZTXWLKPZAV-UHFFFAOYSA-N N-(2-oxo-3-thiolanyl)acetamide Chemical compound CC(=O)NC1CCSC1=O NRFJZTXWLKPZAV-UHFFFAOYSA-N 0.000 description 2
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 2
- OLMDPVRISKOBIT-UHFFFAOYSA-N N-methylcrotsparine Natural products COc1cc2CCN(C)C3c2c(CC34C=CC(=O)C=C4)c1O OLMDPVRISKOBIT-UHFFFAOYSA-N 0.000 description 2
- DDUHZTYCFQRHIY-UHFFFAOYSA-N Negwer: 6874 Natural products COC1=CC(=O)CC(C)C11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 DDUHZTYCFQRHIY-UHFFFAOYSA-N 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- MITFXPHMIHQXPI-UHFFFAOYSA-N Oraflex Chemical compound N=1C2=CC(C(C(O)=O)C)=CC=C2OC=1C1=CC=C(Cl)C=C1 MITFXPHMIHQXPI-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- 102000004211 Platelet factor 4 Human genes 0.000 description 2
- 108090000778 Platelet factor 4 Proteins 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 2
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- 102000019197 Superoxide Dismutase Human genes 0.000 description 2
- 108010012715 Superoxide dismutase Proteins 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical group OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 2
- 108010031372 Tissue Inhibitor of Metalloproteinase-2 Proteins 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- 108090000901 Transferrin Proteins 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 2
- 108091008605 VEGF receptors Proteins 0.000 description 2
- 108010000134 Vascular Cell Adhesion Molecule-1 Proteins 0.000 description 2
- 102000016549 Vascular Endothelial Growth Factor Receptor-2 Human genes 0.000 description 2
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 102100023543 Vascular cell adhesion protein 1 Human genes 0.000 description 2
- OIRDTQYFTABQOQ-UHTZMRCNSA-N Vidarabine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O OIRDTQYFTABQOQ-UHTZMRCNSA-N 0.000 description 2
- DOMHWKQEPDYUQX-LJAQBGIBSA-N [(2r,3r,4s,6r)-3-[(2s,4s,5r,6r)-5-[(2s,4s,5r,6r)-4,5-diformyloxy-6-methyloxan-2-yl]oxy-4-formyloxy-6-methyloxan-2-yl]oxy-6-[[(3s,5r,8r,9s,10s,13r,14s,16s,17r)-16-formyloxy-14-hydroxy-10,13-dimethyl-17-(5-oxo-2h-furan-3-yl)-1,2,3,4,5,6,7,8,9,11,12,15,16,17 Chemical compound C1[C@H](OC=O)[C@H](OC=O)[C@@H](C)O[C@H]1O[C@H]1[C@@H](OC=O)C[C@H](O[C@H]2[C@H](C[C@@H](O[C@@H]2C)O[C@@H]2C[C@@H]3[C@]([C@@H]4[C@H]([C@]5(C[C@@H]([C@@H]([C@@]5(C)CC4)C=4COC(=O)C=4)OC=O)O)CC3)(C)CC2)OC=O)O[C@@H]1C DOMHWKQEPDYUQX-LJAQBGIBSA-N 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 230000001800 adrenalinergic effect Effects 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- NIGUVXFURDGQKZ-UQTBNESHSA-N alpha-Neup5Ac-(2->3)-beta-D-Galp-(1->4)-[alpha-L-Fucp-(1->3)]-beta-D-GlcpNAc Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@H]2[C@@H]([C@@H](O[C@]3(O[C@H]([C@H](NC(C)=O)[C@@H](O)C3)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O)[C@@H](CO)O2)O)[C@@H](CO)O[C@@H](O)[C@@H]1NC(C)=O NIGUVXFURDGQKZ-UQTBNESHSA-N 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- YVPYQUNUQOZFHG-UHFFFAOYSA-N amidotrizoic acid Chemical compound CC(=O)NC1=C(I)C(NC(C)=O)=C(I)C(C(O)=O)=C1I YVPYQUNUQOZFHG-UHFFFAOYSA-N 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 229960001301 amobarbital Drugs 0.000 description 2
- 229960003022 amoxicillin Drugs 0.000 description 2
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 description 2
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 2
- 229960003942 amphotericin b Drugs 0.000 description 2
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 2
- 229960000723 ampicillin Drugs 0.000 description 2
- 229960001220 amsacrine Drugs 0.000 description 2
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 2
- 238000004873 anchoring Methods 0.000 description 2
- 239000002870 angiogenesis inducing agent Substances 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- NWGGKKGAFZIVBJ-UHFFFAOYSA-N antrafenine Chemical compound FC(F)(F)C1=CC=CC(N2CCN(CCOC(=O)C=3C(=CC=CC=3)NC=3C4=CC=C(C=C4N=CC=3)C(F)(F)F)CC2)=C1 NWGGKKGAFZIVBJ-UHFFFAOYSA-N 0.000 description 2
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 229960003121 arginine Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 2
- 229960002274 atenolol Drugs 0.000 description 2
- 230000003143 atherosclerotic effect Effects 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 229960002699 bacampicillin Drugs 0.000 description 2
- PFOLLRNADZZWEX-FFGRCDKISA-N bacampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)[C@H](C(S3)(C)C)C(=O)OC(C)OC(=O)OCC)=CC=CC=C1 PFOLLRNADZZWEX-FFGRCDKISA-N 0.000 description 2
- OGBUMNBNEWYMNJ-UHFFFAOYSA-N batilol Chemical compound CCCCCCCCCCCCCCCCCCOCC(O)CO OGBUMNBNEWYMNJ-UHFFFAOYSA-N 0.000 description 2
- 229950000210 beclometasone dipropionate Drugs 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- BLFLLBZGZJTVJG-UHFFFAOYSA-N benzocaine Chemical compound CCOC(=O)C1=CC=C(N)C=C1 BLFLLBZGZJTVJG-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- ISAOCJYIOMOJEB-UHFFFAOYSA-N benzoin Chemical compound C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 description 2
- CNBGNNVCVSKAQZ-UHFFFAOYSA-N benzydamine Chemical compound C12=CC=CC=C2C(OCCCN(C)C)=NN1CC1=CC=CC=C1 CNBGNNVCVSKAQZ-UHFFFAOYSA-N 0.000 description 2
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 2
- 229960002537 betamethasone Drugs 0.000 description 2
- 230000001588 bifunctional effect Effects 0.000 description 2
- 108091008324 binding proteins Proteins 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 239000012620 biological material Substances 0.000 description 2
- 239000012503 blood component Substances 0.000 description 2
- 210000001218 blood-brain barrier Anatomy 0.000 description 2
- 230000036760 body temperature Effects 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 229960003150 bupivacaine Drugs 0.000 description 2
- 229960002092 busulfan Drugs 0.000 description 2
- 229960001948 caffeine Drugs 0.000 description 2
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 2
- 229960004015 calcitonin Drugs 0.000 description 2
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 2
- 229960000830 captopril Drugs 0.000 description 2
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 229960005361 cefaclor Drugs 0.000 description 2
- QYIYFLOTGYLRGG-GPCCPHFNSA-N cefaclor Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3C(=C(Cl)CS[C@@H]32)C(O)=O)=O)N)=CC=CC=C1 QYIYFLOTGYLRGG-GPCCPHFNSA-N 0.000 description 2
- 229960004841 cefadroxil Drugs 0.000 description 2
- NBFNMSULHIODTC-CYJZLJNKSA-N cefadroxil monohydrate Chemical compound O.C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=C(O)C=C1 NBFNMSULHIODTC-CYJZLJNKSA-N 0.000 description 2
- 229960003012 cefamandole Drugs 0.000 description 2
- OLVCFLKTBJRLHI-AXAPSJFSSA-N cefamandole Chemical compound CN1N=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)[C@H](O)C=3C=CC=CC=3)[C@H]2SC1 OLVCFLKTBJRLHI-AXAPSJFSSA-N 0.000 description 2
- 229960002588 cefradine Drugs 0.000 description 2
- 229940106164 cephalexin Drugs 0.000 description 2
- ZAIPMKNFIOOWCQ-UEKVPHQBSA-N cephalexin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=CC=C1 ZAIPMKNFIOOWCQ-UEKVPHQBSA-N 0.000 description 2
- RDLPVSKMFDYCOR-UEKVPHQBSA-N cephradine Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CCC=CC1 RDLPVSKMFDYCOR-UEKVPHQBSA-N 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- RNFNDJAIBTYOQL-UHFFFAOYSA-N chloral hydrate Chemical compound OC(O)C(Cl)(Cl)Cl RNFNDJAIBTYOQL-UHFFFAOYSA-N 0.000 description 2
- 229960002327 chloral hydrate Drugs 0.000 description 2
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 2
- 229960005091 chloramphenicol Drugs 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 2
- 229960002023 chloroprocaine Drugs 0.000 description 2
- 229960003677 chloroquine Drugs 0.000 description 2
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 2
- QBPFLULOKWLNNW-UHFFFAOYSA-N chrysazin Chemical compound O=C1C2=CC=CC(O)=C2C(=O)C2=C1C=CC=C2O QBPFLULOKWLNNW-UHFFFAOYSA-N 0.000 description 2
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004753 citiolone Drugs 0.000 description 2
- 229960002227 clindamycin Drugs 0.000 description 2
- KDLRVYVGXIQJDK-AWPVFWJPSA-N clindamycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@H](C)Cl)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 KDLRVYVGXIQJDK-AWPVFWJPSA-N 0.000 description 2
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 2
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 2
- 229960004126 codeine Drugs 0.000 description 2
- 239000000084 colloidal system Substances 0.000 description 2
- 229940039231 contrast media Drugs 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 229960003290 cortisone acetate Drugs 0.000 description 2
- 235000000639 cyanocobalamin Nutrition 0.000 description 2
- 239000011666 cyanocobalamin Substances 0.000 description 2
- 229960004244 cyclacillin Drugs 0.000 description 2
- HGBLNBBNRORJKI-WCABBAIRSA-N cyclacillin Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)C1(N)CCCCC1 HGBLNBBNRORJKI-WCABBAIRSA-N 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- QMNFFXRFOJIOKZ-UHFFFAOYSA-N cycloguanil Chemical compound CC1(C)N=C(N)N=C(N)N1C1=CC=C(Cl)C=C1 QMNFFXRFOJIOKZ-UHFFFAOYSA-N 0.000 description 2
- 102000003675 cytokine receptors Human genes 0.000 description 2
- 108010057085 cytokine receptors Proteins 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- 229960000860 dapsone Drugs 0.000 description 2
- GAHOBHHMYUYJDT-UHFFFAOYSA-N desaspidin Chemical compound O=C1C(C(=O)CCC)=C(O)C(C)(C)C(O)=C1CC1=C(O)C(C(=O)CCC)=C(O)C=C1OC GAHOBHHMYUYJDT-UHFFFAOYSA-N 0.000 description 2
- HGKAMARNFGKMLC-RBUKOAKNSA-N dexoxadrol Chemical compound C([C@H]1[C@@H]2OC(OC2)(C=2C=CC=CC=2)C=2C=CC=CC=2)CCCN1 HGKAMARNFGKMLC-RBUKOAKNSA-N 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 229960005223 diatrizoic acid Drugs 0.000 description 2
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 2
- YFAGHNZHGGCZAX-JKIFEVAISA-N dicloxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(Cl)C=CC=C1Cl YFAGHNZHGGCZAX-JKIFEVAISA-N 0.000 description 2
- 229960001585 dicloxacillin Drugs 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 229960000616 diflunisal Drugs 0.000 description 2
- HUPFGZXOMWLGNK-UHFFFAOYSA-N diflunisal Chemical compound C1=C(O)C(C(=O)O)=CC(C=2C(=CC(F)=CC=2)F)=C1 HUPFGZXOMWLGNK-UHFFFAOYSA-N 0.000 description 2
- 229960000648 digitoxin Drugs 0.000 description 2
- WDJUZGPOPHTGOT-XUDUSOBPSA-N digitoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)CC5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O WDJUZGPOPHTGOT-XUDUSOBPSA-N 0.000 description 2
- 229960005156 digoxin Drugs 0.000 description 2
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 2
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 2
- ASXBYYWOLISCLQ-HZYVHMACSA-N dihydrostreptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](CO)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O ASXBYYWOLISCLQ-HZYVHMACSA-N 0.000 description 2
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 2
- 229960004166 diltiazem Drugs 0.000 description 2
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- AUZONCFQVSMFAP-UHFFFAOYSA-N disulfiram Chemical compound CCN(CC)C(=S)SSC(=S)N(CC)CC AUZONCFQVSMFAP-UHFFFAOYSA-N 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 2
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 description 2
- 230000009881 electrostatic interaction Effects 0.000 description 2
- 229960000873 enalapril Drugs 0.000 description 2
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 2
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- SNFOERUNNSHUGP-ZXZARUISSA-N erythrityl tetranitrate Chemical compound [O-][N+](=O)OC[C@@H](O[N+]([O-])=O)[C@@H](O[N+]([O-])=O)CO[N+]([O-])=O SNFOERUNNSHUGP-ZXZARUISSA-N 0.000 description 2
- 229960003276 erythromycin Drugs 0.000 description 2
- 229960005309 estradiol Drugs 0.000 description 2
- 229930182833 estradiol Natural products 0.000 description 2
- 229960001842 estramustine Drugs 0.000 description 2
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 2
- 229960000285 ethambutol Drugs 0.000 description 2
- 229960004447 ethchlorvynol Drugs 0.000 description 2
- 229940052303 ethers for general anesthesia Drugs 0.000 description 2
- GXRZIMHKGDIBEW-UHFFFAOYSA-N ethinamate Chemical compound NC(=O)OC1(C#C)CCCCC1 GXRZIMHKGDIBEW-UHFFFAOYSA-N 0.000 description 2
- 229960002209 ethinamate Drugs 0.000 description 2
- AEOCXXJPGCBFJA-UHFFFAOYSA-N ethionamide Chemical compound CCC1=CC(C(N)=S)=CC=N1 AEOCXXJPGCBFJA-UHFFFAOYSA-N 0.000 description 2
- 229960002001 ethionamide Drugs 0.000 description 2
- 229960003976 etidocaine Drugs 0.000 description 2
- 229960004695 etilefrine Drugs 0.000 description 2
- 229960001690 etomidate Drugs 0.000 description 2
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical compound CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- RRAFCDWBNXTKKO-UHFFFAOYSA-N eugenol Chemical compound COC1=CC(CC=C)=CC=C1O RRAFCDWBNXTKKO-UHFFFAOYSA-N 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 2
- 229940012952 fibrinogen Drugs 0.000 description 2
- 230000003480 fibrinolytic effect Effects 0.000 description 2
- 238000007667 floating Methods 0.000 description 2
- 229960004413 flucytosine Drugs 0.000 description 2
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 2
- SYWHXTATXSMDSB-GSLJADNHSA-N fludrocortisone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)C[C@@H]2O SYWHXTATXSMDSB-GSLJADNHSA-N 0.000 description 2
- 229960000676 flunisolide Drugs 0.000 description 2
- 229960003336 fluorocortisol acetate Drugs 0.000 description 2
- 229960002074 flutamide Drugs 0.000 description 2
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 2
- 229940014144 folate Drugs 0.000 description 2
- 229960000304 folic acid Drugs 0.000 description 2
- 229960000936 fumagillin Drugs 0.000 description 2
- NGGMYCMLYOUNGM-CSDLUJIJSA-N fumagillin Chemical compound C([C@H]([C@H]([C@@H]1[C@]2(C)[C@H](O2)CC=C(C)C)OC)OC(=O)\C=C\C=C\C=C\C=C\C(O)=O)C[C@@]21CO2 NGGMYCMLYOUNGM-CSDLUJIJSA-N 0.000 description 2
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- ZSCRTFONTNMQBL-NTUHNPAUSA-N geranylhydroquinone Chemical compound CC(C)=CCC\C(C)=C\CC1=CC(O)=CC=C1O ZSCRTFONTNMQBL-NTUHNPAUSA-N 0.000 description 2
- 229950004093 geroquinol Drugs 0.000 description 2
- 229950007273 gestaclone Drugs 0.000 description 2
- VUWYSFAIXUWQRQ-VMKBGRNBSA-N gestaclone Chemical compound C1=C(Cl)C2=CC(=O)[C@@H]3C[C@@H]3[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3C[C@@]3(C(=O)C)[C@@]1(C)CC2 VUWYSFAIXUWQRQ-VMKBGRNBSA-N 0.000 description 2
- 229950002795 gestadienol Drugs 0.000 description 2
- SIGSPDASOTUPFS-XUDSTZEESA-N gestodene Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](CC)([C@](C=C4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 SIGSPDASOTUPFS-XUDSTZEESA-N 0.000 description 2
- 229960005352 gestodene Drugs 0.000 description 2
- 229960003812 gestonorone caproate Drugs 0.000 description 2
- 229960004761 gestrinone Drugs 0.000 description 2
- 229950009304 giparmen Drugs 0.000 description 2
- GZZJHPZDXZCDDA-MBJUQXSJSA-N gitaloxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(C[C@@H]([C@@H]([C@@]6(C)CC5)C=5COC(=O)C=5)OC=O)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O GZZJHPZDXZCDDA-MBJUQXSJSA-N 0.000 description 2
- 229950001952 gitaloxin Drugs 0.000 description 2
- 229960001486 gitoformate Drugs 0.000 description 2
- 229960001650 glafenine Drugs 0.000 description 2
- GWOFUCIGLDBNKM-UHFFFAOYSA-N glafenine Chemical compound OCC(O)COC(=O)C1=CC=CC=C1NC1=CC=NC2=CC(Cl)=CC=C12 GWOFUCIGLDBNKM-UHFFFAOYSA-N 0.000 description 2
- PNJUPRNTSWJWAX-ZDUSSCGKSA-N glaziovine Chemical compound C([C@@H]1N(C)CCC=2C=C(C(=C3C=21)O)OC)C13C=CC(=O)C=C1 PNJUPRNTSWJWAX-ZDUSSCGKSA-N 0.000 description 2
- 229950005847 glaziovine Drugs 0.000 description 2
- 229930004040 glaziovine Natural products 0.000 description 2
- 229950004153 gliamilide Drugs 0.000 description 2
- 229960001764 glibornuride Drugs 0.000 description 2
- RMTYNAPTNBJHQI-LLDVTBCESA-N glibornuride Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)N[C@H]1[C@H](C2(C)C)CC[C@@]2(C)[C@H]1O RMTYNAPTNBJHQI-LLDVTBCESA-N 0.000 description 2
- 229950002690 glibutimine Drugs 0.000 description 2
- 229950001623 glicaramide Drugs 0.000 description 2
- 229950000063 glicetanile Drugs 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 229960002972 glutethimide Drugs 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 229940096919 glycogen Drugs 0.000 description 2
- DDUHZTYCFQRHIY-RBHXEPJQSA-N griseofulvin Chemical compound COC1=CC(=O)C[C@@H](C)[C@@]11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 DDUHZTYCFQRHIY-RBHXEPJQSA-N 0.000 description 2
- 229960002867 griseofulvin Drugs 0.000 description 2
- URJHVPKUWOUENU-UHFFFAOYSA-N hadacidin Chemical compound O=CN(O)CC(O)=O URJHVPKUWOUENU-UHFFFAOYSA-N 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 229960003160 hyaluronic acid Drugs 0.000 description 2
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 238000009169 immunotherapy Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 208000017169 kidney disease Diseases 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 229960004616 medroxyprogesterone Drugs 0.000 description 2
- FRQMUZJSZHZSGN-HBNHAYAOSA-N medroxyprogesterone Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](O)(C(C)=O)CC[C@H]21 FRQMUZJSZHZSGN-HBNHAYAOSA-N 0.000 description 2
- INWLQCZOYSRPNW-UHFFFAOYSA-N mepivacaine Chemical compound CN1CCCCC1C(=O)NC1=C(C)C=CC=C1C INWLQCZOYSRPNW-UHFFFAOYSA-N 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 239000003094 microcapsule Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- LXOXCADHKCVBOZ-UHFFFAOYSA-N n-(5-chloro-2-methoxyphenyl)-2-[4-[[5-(2-methylpropyl)pyrimidin-2-yl]sulfamoyl]phenyl]acetamide Chemical compound COC1=CC=C(Cl)C=C1NC(=O)CC1=CC=C(S(=O)(=O)NC=2N=CC(CC(C)C)=CN=2)C=C1 LXOXCADHKCVBOZ-UHFFFAOYSA-N 0.000 description 2
- MQQJZPWSYVPWPF-QYZOEREBSA-N n-[2-[1-[[(1r,4r,5r)-5-bicyclo[2.2.1]hept-2-enyl]methylcarbamoylsulfamoyl]piperidin-4-yl]ethyl]-2-methoxypyridine-3-carboxamide Chemical compound COC1=NC=CC=C1C(=O)NCCC1CCN(S(=O)(=O)NC(=O)NC[C@H]2[C@@H]3C[C@@H](C=C3)C2)CC1 MQQJZPWSYVPWPF-QYZOEREBSA-N 0.000 description 2
- NFGPIRVQFCRUFC-UHFFFAOYSA-N n-[2-[4-(3-cyclohex-3-en-1-yl-2-iminoimidazolidin-1-yl)sulfonylphenyl]ethyl]butanamide Chemical compound C1=CC(CCNC(=O)CCC)=CC=C1S(=O)(=O)N1C(=N)N(C2CC=CCC2)CC1 NFGPIRVQFCRUFC-UHFFFAOYSA-N 0.000 description 2
- DIBGLVNSPMMGHO-UHFFFAOYSA-N n-[2-[4-(cyclohexylcarbamoylsulfamoyl)phenyl]ethyl]-1-ethyl-3-methyl-4-(3-methylbutoxy)pyrazolo[3,4-b]pyridine-5-carboxamide Chemical compound C=1N=C2N(CC)N=C(C)C2=C(OCCC(C)C)C=1C(=O)NCCC(C=C1)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 DIBGLVNSPMMGHO-UHFFFAOYSA-N 0.000 description 2
- 230000001338 necrotic effect Effects 0.000 description 2
- IDBIFFKSXLYUOT-UHFFFAOYSA-N netropsin Chemical compound C1=C(C(=O)NCCC(N)=N)N(C)C=C1NC(=O)C1=CC(NC(=O)CN=C(N)N)=CN1C IDBIFFKSXLYUOT-UHFFFAOYSA-N 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229940127264 non-peptide agonist Drugs 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 239000012038 nucleophile Substances 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 229920001542 oligosaccharide Polymers 0.000 description 2
- 150000002482 oligosaccharides Chemical class 0.000 description 2
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 2
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 2
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 230000010363 phase shift Effects 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 102000036213 phospholipid binding proteins Human genes 0.000 description 2
- 108091011000 phospholipid binding proteins Proteins 0.000 description 2
- ZJAOAACCNHFJAH-UHFFFAOYSA-N phosphonoformic acid Chemical compound OC(=O)P(O)(O)=O ZJAOAACCNHFJAH-UHFFFAOYSA-N 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229950010765 pivalate Drugs 0.000 description 2
- IUGYQRQAERSCNH-UHFFFAOYSA-M pivalate Chemical compound CC(C)(C)C([O-])=O IUGYQRQAERSCNH-UHFFFAOYSA-M 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 150000003180 prostaglandins Chemical class 0.000 description 2
- 235000021251 pulses Nutrition 0.000 description 2
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 2
- AUJXLBOHYWTPFV-UHFFFAOYSA-N quinomycin A Natural products CN1C(=O)C(C)NC(=O)C(NC(=O)C=2N=C3C=CC=CC3=NC=2)COC(=O)C(C(C)C)N(C)C(=O)C2N(C)C(=O)C(C)NC(=O)C(NC(=O)C=3N=C4C=CC=CC4=NC=3)COC(=O)C(C(C)C)N(C)C(=O)C1CSC2SC AUJXLBOHYWTPFV-UHFFFAOYSA-N 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 239000002151 riboflavin Substances 0.000 description 2
- 235000019192 riboflavin Nutrition 0.000 description 2
- 229960002477 riboflavin Drugs 0.000 description 2
- 238000010187 selection method Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 229940063675 spermine Drugs 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 150000005846 sugar alcohols Chemical class 0.000 description 2
- 229960005314 suramin Drugs 0.000 description 2
- FIAFUQMPZJWCLV-UHFFFAOYSA-N suramin Chemical compound OS(=O)(=O)C1=CC(S(O)(=O)=O)=C2C(NC(=O)C3=CC=C(C(=C3)NC(=O)C=3C=C(NC(=O)NC=4C=C(C=CC=4)C(=O)NC=4C(=CC=C(C=4)C(=O)NC=4C5=C(C=C(C=C5C(=CC=4)S(O)(=O)=O)S(O)(=O)=O)S(O)(=O)=O)C)C=CC=3)C)=CC=C(S(O)(=O)=O)C2=C1 FIAFUQMPZJWCLV-UHFFFAOYSA-N 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 229920001059 synthetic polymer Polymers 0.000 description 2
- 229960003433 thalidomide Drugs 0.000 description 2
- 229940126585 therapeutic drug Drugs 0.000 description 2
- 150000007970 thio esters Chemical group 0.000 description 2
- 150000003568 thioethers Chemical group 0.000 description 2
- CNHYKKNIIGEXAY-UHFFFAOYSA-N thiolan-2-imine Chemical compound N=C1CCCS1 CNHYKKNIIGEXAY-UHFFFAOYSA-N 0.000 description 2
- RUVINXPYWBROJD-ONEGZZNKSA-N trans-anethole Chemical compound COC1=CC=C(\C=C\C)C=C1 RUVINXPYWBROJD-ONEGZZNKSA-N 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- 238000000101 transmission high energy electron diffraction Methods 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 229960005356 urokinase Drugs 0.000 description 2
- 210000005167 vascular cell Anatomy 0.000 description 2
- 229960001722 verapamil Drugs 0.000 description 2
- 229960003636 vidarabine Drugs 0.000 description 2
- 108010047303 von Willebrand Factor Proteins 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 229910052724 xenon Inorganic materials 0.000 description 2
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- DBGIVFWFUFKIQN-VIFPVBQESA-N (+)-Fenfluramine Chemical compound CCN[C@@H](C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-VIFPVBQESA-N 0.000 description 1
- GXFZCDMWGMFGFL-KKXMJGKMSA-N (+)-Tubocurarine chloride hydrochloride Chemical compound [Cl-].[Cl-].C([C@H]1[N+](C)(C)CCC=2C=C(C(=C(OC3=CC=C(C=C3)C[C@H]3C=4C=C(C(=CC=4CC[NH+]3C)OC)O3)C=21)O)OC)C1=CC=C(O)C3=C1 GXFZCDMWGMFGFL-KKXMJGKMSA-N 0.000 description 1
- PFTAWBLQPZVEMU-DZGCQCFKSA-N (+)-catechin Chemical compound C1([C@H]2OC3=CC(O)=CC(O)=C3C[C@@H]2O)=CC=C(O)C(O)=C1 PFTAWBLQPZVEMU-DZGCQCFKSA-N 0.000 description 1
- YQYVFVRQLZMJKJ-JBBXEZCESA-N (+)-cyclazocine Chemical compound C([C@@]1(C)C2=CC(O)=CC=C2C[C@@H]2[C@@H]1C)CN2CC1CC1 YQYVFVRQLZMJKJ-JBBXEZCESA-N 0.000 description 1
- DNXHEGUUPJUMQT-UHFFFAOYSA-N (+)-estrone Natural products OC1=CC=C2C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 DNXHEGUUPJUMQT-UHFFFAOYSA-N 0.000 description 1
- ZCVAOQKBXKSDMS-PVAVHDDUSA-N (+)-trans-(S)-allethrin Chemical compound CC1(C)[C@H](C=C(C)C)[C@H]1C(=O)O[C@@H]1C(C)=C(CC=C)C(=O)C1 ZCVAOQKBXKSDMS-PVAVHDDUSA-N 0.000 description 1
- DBGIVFWFUFKIQN-UHFFFAOYSA-N (+-)-Fenfluramine Chemical compound CCNC(C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-UHFFFAOYSA-N 0.000 description 1
- JWZZKOKVBUJMES-UHFFFAOYSA-N (+-)-Isoprenaline Chemical compound CC(C)NCC(O)C1=CC=C(O)C(O)=C1 JWZZKOKVBUJMES-UHFFFAOYSA-N 0.000 description 1
- AYIRNRDRBQJXIF-NXEZZACHSA-N (-)-Florfenicol Chemical compound CS(=O)(=O)C1=CC=C([C@@H](O)[C@@H](CF)NC(=O)C(Cl)Cl)C=C1 AYIRNRDRBQJXIF-NXEZZACHSA-N 0.000 description 1
- UIKROCXWUNQSPJ-VIFPVBQESA-N (-)-cotinine Chemical compound C1CC(=O)N(C)[C@@H]1C1=CC=CN=C1 UIKROCXWUNQSPJ-VIFPVBQESA-N 0.000 description 1
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- YIUNVJGEMXODJX-UHFFFAOYSA-M (1-benzylpyridin-1-ium-3-yl) n,n-dimethylcarbamate;bromide Chemical compound [Br-].CN(C)C(=O)OC1=CC=C[N+](CC=2C=CC=CC=2)=C1 YIUNVJGEMXODJX-UHFFFAOYSA-M 0.000 description 1
- XEHKKWZHSSPBNZ-UHFFFAOYSA-M (1-ethyl-1-methylpiperidin-1-ium-4-yl) 2-(azepan-1-yl)-2-(1,2-benzoxazol-3-yl)acetate;iodide Chemical compound [I-].C1C[N+](CC)(C)CCC1OC(=O)C(C=1C2=CC=CC=C2ON=1)N1CCCCCC1 XEHKKWZHSSPBNZ-UHFFFAOYSA-M 0.000 description 1
- HZVNJIOPUIMLRO-UHFFFAOYSA-M (1-ethyl-1-methylpyrrolidin-1-ium-3-yl) 2-cyclopentyl-2-phenylacetate;bromide Chemical compound [Br-].C1[N+](CC)(C)CCC1OC(=O)C(C=1C=CC=CC=1)C1CCCC1 HZVNJIOPUIMLRO-UHFFFAOYSA-M 0.000 description 1
- RBKLLGUTSKKRLX-UHFFFAOYSA-M (1-methyl-1-azoniabicyclo[2.2.2]octan-3-yl) 2-cyclohexyl-2-hydroxy-2-phenylacetate;bromide Chemical compound [Br-].C1C[N+](C)(C2)CCC1C2OC(=O)C(O)(C=1C=CC=CC=1)C1CCCCC1 RBKLLGUTSKKRLX-UHFFFAOYSA-M 0.000 description 1
- JAXUFJQVMKFWNB-UHFFFAOYSA-N (1-methylpyrrolidin-2-yl)methyl 2,2-bis(4-chlorophenoxy)acetate Chemical compound CN1CCCC1COC(=O)C(OC=1C=CC(Cl)=CC=1)OC1=CC=C(Cl)C=C1 JAXUFJQVMKFWNB-UHFFFAOYSA-N 0.000 description 1
- VBQROPPRMFZXNC-NBVRZTHBSA-N (11e)-11-[2-(4-methylpiperazin-1-yl)-2-oxoethylidene]-5h-benzo[c][1]benzazepin-6-one Chemical compound C1CN(C)CCN1C(=O)\C=C\1C2=CC=CC=C2C(=O)NC2=CC=CC=C2/1 VBQROPPRMFZXNC-NBVRZTHBSA-N 0.000 description 1
- WFTSRDISOMSAQC-ZNFOTRSXSA-N (1R,15S,17R,18R,19S,20S)-3-[2-(diethylamino)ethyl]-6,18-dimethoxy-17-[oxo-(3,4,5-trimethoxyphenyl)methoxy]-11,12,14,15,16,17,18,19,20,21-decahydro-1H-yohimban-19-carboxylic acid methyl ester Chemical compound O([C@@H]1C[C@H]2[C@@H]([C@@H]([C@H]1OC)C(=O)OC)C[C@@H]1C=3N(C4=CC(OC)=CC=C4C=3CCN1C2)CCN(CC)CC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 WFTSRDISOMSAQC-ZNFOTRSXSA-N 0.000 description 1
- DAASOABUJRMZAD-NRYKZSQYSA-N (1R,4S,5S)-5-(bromomethyl)-1,2,3,4,7,7-hexachlorobicyclo[2.2.1]hept-2-ene Chemical compound BrC[C@H]1C[C@@]2(Cl)C(Cl)=C(Cl)[C@]1(Cl)C2(Cl)Cl DAASOABUJRMZAD-NRYKZSQYSA-N 0.000 description 1
- VCZPUGSOJXZKIP-YPMHNXCESA-N (1S,2R)-cicloxilic acid Chemical compound OC(=O)[C@H]1CCCC[C@]1(O)C1=CC=CC=C1 VCZPUGSOJXZKIP-YPMHNXCESA-N 0.000 description 1
- AFZOCGNTFCGOEE-QUCCMNQESA-N (1S,9R)-10-(cyclopropylmethyl)-1-ethyl-13,13-dimethyl-10-azatricyclo[7.3.1.02,7]trideca-2(7),3,5-trien-4-ol Chemical compound C([C@]1(C2=CC(O)=CC=C2C[C@@H]2C1(C)C)CC)CN2CC1CC1 AFZOCGNTFCGOEE-QUCCMNQESA-N 0.000 description 1
- SMZVRZPJXBGNFT-LBPRGKRZSA-N (1r)-1-(1,3-dioxan-5-yl)-n,n-dimethyl-1-pyridin-3-ylmethanamine Chemical compound C1([C@@H](N(C)C)C=2C=NC=CC=2)COCOC1 SMZVRZPJXBGNFT-LBPRGKRZSA-N 0.000 description 1
- ZNMXKJDERFMXNF-ZETCQYMHSA-N (1r)-1-(2-hydroxy-1,3,2-benzodioxaborol-5-yl)-2-(methylamino)ethanol Chemical compound CNC[C@H](O)C1=CC=C2OB(O)OC2=C1 ZNMXKJDERFMXNF-ZETCQYMHSA-N 0.000 description 1
- SSEBTPPFLLCUMN-CYBMUJFWSA-N (1r)-2-(tert-butylamino)-1-(7-ethyl-1-benzofuran-2-yl)ethanol Chemical compound CCC1=CC=CC2=C1OC([C@H](O)CNC(C)(C)C)=C2 SSEBTPPFLLCUMN-CYBMUJFWSA-N 0.000 description 1
- BXAMVNHBXFXFSK-LXTVHRRPSA-N (1r,2r,3r,4s)-2-n,2-n,3-n,3-n-tetraethylbicyclo[2.2.1]hept-5-ene-2,3-dicarboxamide Chemical compound C1[C@@H]2C=C[C@H]1[C@@H](C(=O)N(CC)CC)[C@@H]2C(=O)N(CC)CC BXAMVNHBXFXFSK-LXTVHRRPSA-N 0.000 description 1
- LUYXWZOOMKBUMB-ONJZCGHCSA-N (1r,4ar,12as)-3-acetyl-1-amino-4,4a,6,7-tetrahydroxy-8,11-dimethyl-12,12a-dihydro-1h-tetracene-2,5-dione Chemical compound C1=C(C)C(O)=C2C(O)=C(C([C@]3(O)C(O)=C(C([C@H](N)[C@@H]3C3)=O)C(=O)C)=O)C3=C(C)C2=C1 LUYXWZOOMKBUMB-ONJZCGHCSA-N 0.000 description 1
- AAFJXZWCNVJTMK-GUCUJZIJSA-N (1s,2r)-1-[(2s)-oxiran-2-yl]-2-[(2r)-oxiran-2-yl]ethane-1,2-diol Chemical compound C([C@@H]1[C@H](O)[C@H](O)[C@H]2OC2)O1 AAFJXZWCNVJTMK-GUCUJZIJSA-N 0.000 description 1
- TWUSDDMONZULSC-QMTHXVAHSA-N (1s,2r)-2-(tert-butylamino)-1-(2,5-dimethoxyphenyl)propan-1-ol Chemical compound COC1=CC=C(OC)C([C@H](O)[C@@H](C)NC(C)(C)C)=C1 TWUSDDMONZULSC-QMTHXVAHSA-N 0.000 description 1
- YATDSXRLIUJOQN-SVRRBLITSA-N (2,3,4,5,6-pentafluorophenyl)methyl (1r,3s)-3-(2,2-dichloroethenyl)-2,2-dimethylcyclopropane-1-carboxylate Chemical compound CC1(C)[C@H](C=C(Cl)Cl)[C@H]1C(=O)OCC1=C(F)C(F)=C(F)C(F)=C1F YATDSXRLIUJOQN-SVRRBLITSA-N 0.000 description 1
- XEDWWPGWIXPVRQ-UHFFFAOYSA-N (2,3,4-trihydroxyphenyl)-(3,4,5-trihydroxyphenyl)methanone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC(O)=C(O)C(O)=C1 XEDWWPGWIXPVRQ-UHFFFAOYSA-N 0.000 description 1
- JCKOFFJWBOVAAV-UHFFFAOYSA-N (2,5-dimethylfuran-3-yl)-(4-hydroxy-3,5-diiodophenyl)methanone Chemical compound O1C(C)=CC(C(=O)C=2C=C(I)C(O)=C(I)C=2)=C1C JCKOFFJWBOVAAV-UHFFFAOYSA-N 0.000 description 1
- FTOLAXZXUUHQQX-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 1-(3-cyano-3,3-diphenylpropyl)-4-phenylpiperidine-4-carboxylate Chemical compound C1CN(CCC(C#N)(C=2C=CC=CC=2)C=2C=CC=CC=2)CCC1(C=1C=CC=CC=1)C(=O)ON1C(=O)CCC1=O FTOLAXZXUUHQQX-UHFFFAOYSA-N 0.000 description 1
- ILKQVTIMOGNSAS-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-(7-amino-4-methyl-2-oxochromen-3-yl)acetate Chemical compound O=C1OC=2C=C(N)C=CC=2C(C)=C1CC(=O)ON1C(=O)CCC1=O ILKQVTIMOGNSAS-UHFFFAOYSA-N 0.000 description 1
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 1
- PVGATNRYUYNBHO-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(2,5-dioxopyrrol-1-yl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O PVGATNRYUYNBHO-UHFFFAOYSA-N 0.000 description 1
- BQWBEDSJTMWJAE-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[(2-iodoacetyl)amino]benzoate Chemical compound C1=CC(NC(=O)CI)=CC=C1C(=O)ON1C(=O)CCC1=O BQWBEDSJTMWJAE-UHFFFAOYSA-N 0.000 description 1
- GKSPIZSKQWTXQG-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[1-(pyridin-2-yldisulfanyl)ethyl]benzoate Chemical compound C=1C=C(C(=O)ON2C(CCC2=O)=O)C=CC=1C(C)SSC1=CC=CC=N1 GKSPIZSKQWTXQG-UHFFFAOYSA-N 0.000 description 1
- PMJWDPGOWBRILU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[4-(2,5-dioxopyrrol-1-yl)phenyl]butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCC(C=C1)=CC=C1N1C(=O)C=CC1=O PMJWDPGOWBRILU-UHFFFAOYSA-N 0.000 description 1
- RBAFCMJBDZWZIV-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-azido-2-hydroxybenzoate Chemical compound OC1=CC(N=[N+]=[N-])=CC=C1C(=O)ON1C(=O)CCC1=O RBAFCMJBDZWZIV-UHFFFAOYSA-N 0.000 description 1
- LWAVGNJLLQSNNN-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-azidobenzoate Chemical compound C1=CC(N=[N+]=[N-])=CC=C1C(=O)ON1C(=O)CCC1=O LWAVGNJLLQSNNN-UHFFFAOYSA-N 0.000 description 1
- FUOJEDZPVVDXHI-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 5-azido-2-nitrobenzoate Chemical compound [O-][N+](=O)C1=CC=C(N=[N+]=[N-])C=C1C(=O)ON1C(=O)CCC1=O FUOJEDZPVVDXHI-UHFFFAOYSA-N 0.000 description 1
- NGXDNMNOQDVTRL-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-(4-azido-2-nitroanilino)hexanoate Chemical compound [O-][N+](=O)C1=CC(N=[N+]=[N-])=CC=C1NCCCCCC(=O)ON1C(=O)CCC1=O NGXDNMNOQDVTRL-UHFFFAOYSA-N 0.000 description 1
- QNIUBYBLEIJTNS-UHFFFAOYSA-N (2-amino-2-oxoethyl) 2-[3-(trifluoromethyl)anilino]benzoate Chemical compound NC(=O)COC(=O)C1=CC=CC=C1NC1=CC=CC(C(F)(F)F)=C1 QNIUBYBLEIJTNS-UHFFFAOYSA-N 0.000 description 1
- XKKGMVDIONCFKP-UHFFFAOYSA-N (2-butyl-4-methoxynaphthalen-1-yl) acetate Chemical compound C1=CC=CC2=C(OC(C)=O)C(CCCC)=CC(OC)=C21 XKKGMVDIONCFKP-UHFFFAOYSA-N 0.000 description 1
- BZHSRUIDLJDTTJ-UHFFFAOYSA-N (2-ethoxy-2-oxoethyl) 3,5-diacetamido-2,4,6-triiodobenzoate Chemical compound CCOC(=O)COC(=O)C1=C(I)C(NC(C)=O)=C(I)C(NC(C)=O)=C1I BZHSRUIDLJDTTJ-UHFFFAOYSA-N 0.000 description 1
- OWCAKKIRPJUQFO-UHFFFAOYSA-N (2-methyl-2-nonyl-1,3-dioxolan-4-yl)methyl carbamate Chemical compound CCCCCCCCCC1(C)OCC(COC(N)=O)O1 OWCAKKIRPJUQFO-UHFFFAOYSA-N 0.000 description 1
- JQWAHKMIYCERGA-UHFFFAOYSA-N (2-nonanoyloxy-3-octadeca-9,12-dienoyloxypropoxy)-[2-(trimethylazaniumyl)ethyl]phosphinate Chemical compound CCCCCCCCC(=O)OC(COP([O-])(=O)CC[N+](C)(C)C)COC(=O)CCCCCCCC=CCC=CCCCCC JQWAHKMIYCERGA-UHFFFAOYSA-N 0.000 description 1
- OILXMJHPFNGGTO-UHFFFAOYSA-N (22E)-(24xi)-24-methylcholesta-5,22-dien-3beta-ol Natural products C1C=C2CC(O)CCC2(C)C2C1C1CCC(C(C)C=CC(C)C(C)C)C1(C)CC2 OILXMJHPFNGGTO-UHFFFAOYSA-N 0.000 description 1
- RQOCXCFLRBRBCS-UHFFFAOYSA-N (22E)-cholesta-5,7,22-trien-3beta-ol Natural products C1C(O)CCC2(C)C(CCC3(C(C(C)C=CCC(C)C)CCC33)C)C3=CC=C21 RQOCXCFLRBRBCS-UHFFFAOYSA-N 0.000 description 1
- DZLOHEOHWICNIL-QGZVFWFLSA-N (2R)-2-[6-(4-chlorophenoxy)hexyl]-2-oxiranecarboxylic acid ethyl ester Chemical compound C=1C=C(Cl)C=CC=1OCCCCCC[C@]1(C(=O)OCC)CO1 DZLOHEOHWICNIL-QGZVFWFLSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- MQHLMHIZUIDKOO-OKZBNKHCSA-N (2R,6S)-2,6-dimethyl-4-[(2S)-2-methyl-3-[4-(2-methylbutan-2-yl)phenyl]propyl]morpholine Chemical compound C1=CC(C(C)(C)CC)=CC=C1C[C@H](C)CN1C[C@@H](C)O[C@@H](C)C1 MQHLMHIZUIDKOO-OKZBNKHCSA-N 0.000 description 1
- QDZOEBFLNHCSSF-PFFBOGFISA-N (2S)-2-[[(2R)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-1-[(2R)-2-amino-5-carbamimidamidopentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-N-[(2R)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]pentanediamide Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CCCNC(N)=N)C1=CC=CC=C1 QDZOEBFLNHCSSF-PFFBOGFISA-N 0.000 description 1
- URCIJDUOBBSMII-HOTGVXAUSA-N (2S)-N-[(2S)-1-phenoxypropan-2-yl]-1-phenylpropan-2-amine Chemical compound C([C@H](C)N[C@@H](C)CC=1C=CC=CC=1)OC1=CC=CC=C1 URCIJDUOBBSMII-HOTGVXAUSA-N 0.000 description 1
- RDVXUHOSYIBGBT-ZWNOBZJWSA-N (2S,3R)-bifluranol Chemical compound C1([C@@H](C)[C@@H](CC)C=2C=C(F)C(O)=CC=2)=CC=C(O)C(F)=C1 RDVXUHOSYIBGBT-ZWNOBZJWSA-N 0.000 description 1
- DHKKONBXGAAFTB-OTYYAQKOSA-N (2e,6e)-2,6-bis[(4-hydroxy-3-methoxyphenyl)methylidene]cyclohexan-1-one Chemical compound C1=C(O)C(OC)=CC(\C=C/2C(C(=C/C=3C=C(OC)C(O)=CC=3)/CCC\2)=O)=C1 DHKKONBXGAAFTB-OTYYAQKOSA-N 0.000 description 1
- MLYCFWZIAJAIGW-LLVKDONJSA-N (2r)-1-(2,5-dimethoxy-4-methylphenyl)butan-2-amine Chemical compound CC[C@@H](N)CC1=CC(OC)=C(C)C=C1OC MLYCFWZIAJAIGW-LLVKDONJSA-N 0.000 description 1
- IYQDIWRBEQWANY-ZECSTPGPSA-N (2r)-1-(tert-butylamino)-3-[(e)-1-cyclopropylethylideneamino]oxypropan-2-ol Chemical compound CC(C)(C)NC[C@@H](O)CO\N=C(/C)C1CC1 IYQDIWRBEQWANY-ZECSTPGPSA-N 0.000 description 1
- JNDJPKHYZWRRIS-MRXNPFEDSA-N (2r)-1-[4-[2-(cyclopropylmethoxy)ethoxy]phenoxy]-3-(propan-2-ylamino)propan-2-ol Chemical compound C1=CC(OC[C@H](O)CNC(C)C)=CC=C1OCCOCC1CC1 JNDJPKHYZWRRIS-MRXNPFEDSA-N 0.000 description 1
- FXPSJTKESYQXLX-HQVZTVAUSA-N (2r)-2-acetamido-3-(2-methyl-3-oxo-3-phenylpropyl)sulfanylpropanoic acid Chemical compound CC(=O)N[C@H](C(O)=O)CSCC(C)C(=O)C1=CC=CC=C1 FXPSJTKESYQXLX-HQVZTVAUSA-N 0.000 description 1
- XEQLFNPSYWZPOW-NUOYRARPSA-N (2r)-4-amino-n-[(1r,2s,3r,4r,5s)-5-amino-4-[(2r,3r,4r,5s,6r)-3-amino-6-(aminomethyl)-4,5-dihydroxyoxan-2-yl]oxy-3-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-2-hydroxycyclohexyl]-2-hydroxybutanamide Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O[C@@H]1[C@@H]([C@H](O)[C@@H](CO)O1)O)O)NC(=O)[C@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1N XEQLFNPSYWZPOW-NUOYRARPSA-N 0.000 description 1
- HBIDWFPFQACMBQ-MUUNZHRXSA-N (2r)-4-benzhydryl-2-[(3,4-dimethoxyphenyl)methyl]-1,2-dimethylpiperazine Chemical compound C1=C(OC)C(OC)=CC=C1C[C@@]1(C)N(C)CCN(C(C=2C=CC=CC=2)C=2C=CC=CC=2)C1 HBIDWFPFQACMBQ-MUUNZHRXSA-N 0.000 description 1
- PAYBYKKERMGTSS-MNCSTQPFSA-N (2r,3r,3as,9ar)-7-fluoro-2-(hydroxymethyl)-6-imino-2,3,3a,9a-tetrahydrofuro[1,2][1,3]oxazolo[3,4-a]pyrimidin-3-ol Chemical compound N=C1C(F)=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 PAYBYKKERMGTSS-MNCSTQPFSA-N 0.000 description 1
- OCFOTEIMZBKQFS-DGMGPCKZSA-N (2r,3r,4s,5s,6r)-2-[(1r,2s,3s,4r,6s)-6-amino-3-[(2s,3r,4s,5s,6r)-4-amino-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-4-[[(2s)-4-amino-2-hydroxybutyl]amino]-2-hydroxycyclohexyl]oxy-6-(aminomethyl)oxane-3,4,5-triol Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O OCFOTEIMZBKQFS-DGMGPCKZSA-N 0.000 description 1
- NGJOOUAZHCZCOW-WNCULLNHSA-N (2r,3s)-2-(2,2-diphenyl-1,3-benzodioxol-5-yl)-3,4-dihydro-2h-chromene-3,5,7-triol Chemical compound O1C2=CC([C@H]3OC4=CC(O)=CC(O)=C4C[C@@H]3O)=CC=C2OC1(C=1C=CC=CC=1)C1=CC=CC=C1 NGJOOUAZHCZCOW-WNCULLNHSA-N 0.000 description 1
- PSHBCUHYCLAGPZ-BAOLWLNASA-N (2r,6r)-6-(3-chlorophenoxy)-2-methyl-1-oxa-4-azaspiro[4.5]decan-3-one Chemical compound N1C(=O)[C@@H](C)OC11[C@H](OC=2C=C(Cl)C=CC=2)CCCC1 PSHBCUHYCLAGPZ-BAOLWLNASA-N 0.000 description 1
- VKCGSTZAZMNTRV-DHUJRADRSA-N (2s)-2,6-bis(hexadecanoylamino)hexanoic acid Chemical compound CCCCCCCCCCCCCCCC(=O)NCCCC[C@@H](C(O)=O)NC(=O)CCCCCCCCCCCCCCC VKCGSTZAZMNTRV-DHUJRADRSA-N 0.000 description 1
- RDJGLLICXDHJDY-NSHDSACASA-N (2s)-2-(3-phenoxyphenyl)propanoic acid Chemical compound OC(=O)[C@@H](C)C1=CC=CC(OC=2C=CC=CC=2)=C1 RDJGLLICXDHJDY-NSHDSACASA-N 0.000 description 1
- VUAFHZCUKUDDBC-SCSAIBSYSA-N (2s)-2-[(2-methyl-2-sulfanylpropanoyl)amino]-3-sulfanylpropanoic acid Chemical compound CC(C)(S)C(=O)N[C@H](CS)C(O)=O VUAFHZCUKUDDBC-SCSAIBSYSA-N 0.000 description 1
- QJERBBQXOMUURJ-INIZCTEOSA-N (2s)-2-[(4-chlorobenzoyl)amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C1=CC=C(Cl)C=C1 QJERBBQXOMUURJ-INIZCTEOSA-N 0.000 description 1
- RJMIEHBSYVWVIN-NSHDSACASA-N (2s)-2-[4-(3-oxo-1h-isoindol-2-yl)phenyl]propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1N1C(=O)C2=CC=CC=C2C1 RJMIEHBSYVWVIN-NSHDSACASA-N 0.000 description 1
- MCEHFIXEKNKSRW-LBPRGKRZSA-N (2s)-2-[[3,5-dichloro-4-[(2,4-diaminopteridin-6-yl)methyl-methylamino]benzoyl]amino]pentanedioic acid Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=C(Cl)C=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1Cl MCEHFIXEKNKSRW-LBPRGKRZSA-N 0.000 description 1
- WKAVKKUXZAWHDM-JEDNCBNOSA-N (2s)-2-acetamidopentanedioic acid;2-(dimethylamino)ethanol Chemical compound CN(C)CCO.CC(=O)N[C@H](C(O)=O)CCC(O)=O WKAVKKUXZAWHDM-JEDNCBNOSA-N 0.000 description 1
- JRBXPUUAYKCCLQ-QMMMGPOBSA-N (2s)-2-amino-2-[3-hydroxy-4-(hydroxymethyl)phenyl]acetic acid Chemical compound OC(=O)[C@@H](N)C1=CC=C(CO)C(O)=C1 JRBXPUUAYKCCLQ-QMMMGPOBSA-N 0.000 description 1
- UYTSRQMXRROFPU-LIIDHCAMSA-N (2s)-2-amino-2-deuterio-3-fluoropropanoic acid Chemical compound FC[C@](N)([2H])C(O)=O UYTSRQMXRROFPU-LIIDHCAMSA-N 0.000 description 1
- RPEPXOHTYVXVMA-CIUDSAMLSA-N (2s)-2-amino-5-[[(2s)-1-[[(1s)-1-carboxy-4-(3h-diazirin-3-yl)-4-oxobutyl]amino]-5-(3h-diazirin-3-yl)-1,5-dioxopentan-2-yl]amino]-5-oxopentanoic acid Chemical compound C([C@H](NC(=O)CC[C@H](N)C(O)=O)C(=O)N[C@@H](CCC(=O)C1N=N1)C(O)=O)CC(=O)C1N=N1 RPEPXOHTYVXVMA-CIUDSAMLSA-N 0.000 description 1
- TZJGKUCHNFFHGN-LURJTMIESA-N (2s)-2-aminohexanal Chemical compound CCCC[C@H](N)C=O TZJGKUCHNFFHGN-LURJTMIESA-N 0.000 description 1
- OBDSVYOSYSKVMX-JTQLQIEISA-N (2s)-n,n-dimethyl-1-phenylpropan-2-amine Chemical compound CN(C)[C@@H](C)CC1=CC=CC=C1 OBDSVYOSYSKVMX-JTQLQIEISA-N 0.000 description 1
- ZALQKBPFOWAOHH-SUMWQHHRSA-N (2s)-n-[(2r)-1-[(2-amino-2-oxoethyl)amino]-1-oxo-2-phenylbutan-2-yl]pyrrolidine-2-carboxamide Chemical compound N([C@@](CC)(C(=O)NCC(N)=O)C=1C=CC=CC=1)C(=O)[C@@H]1CCCN1 ZALQKBPFOWAOHH-SUMWQHHRSA-N 0.000 description 1
- KYHUYMLIVQFXRI-XYUQHQMCSA-N (2s)-n-[(2r)-1-[[(3s,6s,8s,12s,13r,16s,17r,20s,23s)-13-[(2s)-butan-2-yl]-12-hydroxy-20-[(4-methoxyphenyl)methyl]-6,17,21-trimethyl-3-(2-methylpropyl)-2,5,7,10,15,19,22-heptaoxo-8-propan-2-yl-9,18-dioxa-1,4,14,21-tetrazabicyclo[21.3.0]hexacosan-16-yl]amino Chemical compound CN([C@H](CC(C)C)C(=O)N[C@@H]1C(=O)N[C@@H]([C@H](CC(=O)O[C@H](C(=O)[C@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N2CCC[C@H]2C(=O)N(C)[C@@H](CC=2C=CC(OC)=CC=2)C(=O)O[C@@H]1C)C(C)C)O)[C@@H](C)CC)C(=O)[C@@H]1CCCN1C(=O)C(C)O KYHUYMLIVQFXRI-XYUQHQMCSA-N 0.000 description 1
- STMUIVWQDNMZCA-CRKDRTNXSA-N (2s,3r,4s,5r)-2-(6-aminopurin-9-yl)-2-fluoro-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@]1(F)O[C@H](CO)[C@@H](O)[C@H]1O STMUIVWQDNMZCA-CRKDRTNXSA-N 0.000 description 1
- LJRDOKAZOAKLDU-UDXJMMFXSA-N (2s,3s,4r,5r,6r)-5-amino-2-(aminomethyl)-6-[(2r,3s,4r,5s)-5-[(1r,2r,3s,5r,6s)-3,5-diamino-2-[(2s,3r,4r,5s,6r)-3-amino-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-hydroxycyclohexyl]oxy-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl]oxyoxane-3,4-diol;sulfuric ac Chemical compound OS(O)(=O)=O.N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO LJRDOKAZOAKLDU-UDXJMMFXSA-N 0.000 description 1
- IPIHZIYZLLMCRF-APMXPKBPSA-N (2s,4as,6ar,6as,6br,8ar,10s,12as,14br)-10-[(1s,2r)-2-carboxycyclohexanecarbonyl]oxy-2,4a,6a,6b,9,9,12a-heptamethyl-13-oxo-3,4,5,6,6a,7,8,8a,10,11,12,14b-dodecahydro-1h-picene-2-carboxylic acid Chemical compound O([C@@H]1C([C@H]2[C@]([C@@H]3[C@@]([C@@]4(CC[C@@]5(C)CC[C@@](C)(C[C@H]5C4=CC3=O)C(O)=O)C)(C)CC2)(C)CC1)(C)C)C(=O)[C@H]1CCCC[C@H]1C(O)=O IPIHZIYZLLMCRF-APMXPKBPSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- BLHZPPIRNRDRSC-HSDAMQNGSA-N (2s,5r,6r)-6-[[(2r)-2-(3,4-dihydroxyphenyl)-2-[(4-ethyl-2,3-dioxopiperazine-1-carbonyl)amino]acetyl]amino]-6-formamido-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid Chemical compound O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=C(O)C(O)=CC=1)C(=O)N[C@]1(NC=O)C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 BLHZPPIRNRDRSC-HSDAMQNGSA-N 0.000 description 1
- FRUAVHAXMXYUGM-NFFDBFGFSA-N (2s,5r,6r)-6-[[(2r)-2-(furan-2-carbonyloxy)-4-methylpentanoyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid Chemical compound O([C@H](CC(C)C)C(=O)N[C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=CC=CO1 FRUAVHAXMXYUGM-NFFDBFGFSA-N 0.000 description 1
- BSIJLJGDLNRGBY-NXWNEQKCSA-N (2s,5r,6r)-6-[[(2r)-2-[[2-(4-chlorophenoxy)-2-methylpropanoyl]amino]-2-phenylacetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid Chemical compound N([C@@H](C(=O)N[C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C=1C=CC=CC=1)C(=O)C(C)(C)OC1=CC=C(Cl)C=C1 BSIJLJGDLNRGBY-NXWNEQKCSA-N 0.000 description 1
- YSUBQYRZZFVMTL-YNDGFOBUSA-N (2s,5r,6r)-6-[[(2r)-2-[[3-[(e)-furan-2-ylmethylideneamino]-2-oxoimidazolidine-1-carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid Chemical compound N([C@@H](C(=O)N[C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C=1C=CC(O)=CC=1)C(=O)N(C1=O)CCN1\N=C\C1=CC=CO1 YSUBQYRZZFVMTL-YNDGFOBUSA-N 0.000 description 1
- DAVPSCAAXXVSFU-ALEPSDHESA-N (2s,5r,6r)-6-bromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid Chemical compound OC(=O)[C@H]1C(C)(C)S[C@@H]2[C@H](Br)C(=O)N21 DAVPSCAAXXVSFU-ALEPSDHESA-N 0.000 description 1
- SRJOJNGMYPRTOL-UHFFFAOYSA-N (3,3,5-trimethylcyclohexyl) 2-hydroxypropanoate Chemical compound CC(O)C(=O)OC1CC(C)CC(C)(C)C1 SRJOJNGMYPRTOL-UHFFFAOYSA-N 0.000 description 1
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 1
- YXZWPKTWLHVLEY-CAOOACKPSA-N (3e)-3-(3-chloro-5,6-dihydrobenzo[c][1]benzazepin-11-ylidene)-n,n-dimethylpropan-1-amine Chemical compound C1NC2=CC(Cl)=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 YXZWPKTWLHVLEY-CAOOACKPSA-N 0.000 description 1
- CMPHKNLTWCBULI-LICLKQGHSA-N (3e)-3-(4-chlorodibenzo[1,2-a:3',2'-e][7]annulen-11-ylidene)-n,n-dimethylpropan-1-amine oxide Chemical compound C1=CC2=C(Cl)C=CC=C2C(=C/CC[N+](C)([O-])C)/C2=CC=CC=C21 CMPHKNLTWCBULI-LICLKQGHSA-N 0.000 description 1
- YFFIQHDMUUCCJP-LDADJPATSA-N (3e)-n,n-dimethyl-3-(5-methyl-6h-benzo[c][1]benzazepin-11-ylidene)propan-1-amine Chemical compound C1N(C)C2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 YFFIQHDMUUCCJP-LDADJPATSA-N 0.000 description 1
- HPWIIERXAFODPP-GHBBWTPBSA-N (3r,4r)-3,6-diamino-n-[(3s,6z,9s,12s,15s)-3-[(6r)-2-amino-1,4,5,6-tetrahydropyrimidin-6-yl]-6-[(carbamoylamino)methylidene]-9,12-bis(hydroxymethyl)-2,5,8,11,14-pentaoxo-1,4,7,10,13-pentazacyclohexadec-15-yl]-4-hydroxyhexanamide Chemical compound N1C(=O)\C(=C\NC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)C[C@@H](N)[C@H](O)CCN)CNC(=O)[C@@H]1[C@@H]1NC(=N)NCC1 HPWIIERXAFODPP-GHBBWTPBSA-N 0.000 description 1
- OILXMJHPFNGGTO-FQRXBOAXSA-N (3s)-17-[(e)-5,6-dimethylhept-3-en-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-ol Chemical compound C1C=C2C[C@@H](O)CCC2(C)C2C1C1CCC(C(C)/C=C/C(C)C(C)C)C1(C)CC2 OILXMJHPFNGGTO-FQRXBOAXSA-N 0.000 description 1
- VCOPTHOUUNAYKQ-WBTCAYNUSA-N (3s)-3,6-diamino-n-[[(2s,5s,8e,11s,15s)-15-amino-11-[(6r)-2-amino-1,4,5,6-tetrahydropyrimidin-6-yl]-8-[(carbamoylamino)methylidene]-2-(hydroxymethyl)-3,6,9,12,16-pentaoxo-1,4,7,10,13-pentazacyclohexadec-5-yl]methyl]hexanamide;(3s)-3,6-diamino-n-[[(2s,5s,8 Chemical compound N1C(=O)\C(=C/NC(N)=O)NC(=O)[C@H](CNC(=O)C[C@@H](N)CCCN)NC(=O)[C@H](C)NC(=O)[C@@H](N)CNC(=O)[C@@H]1[C@@H]1NC(N)=NCC1.N1C(=O)\C(=C/NC(N)=O)NC(=O)[C@H](CNC(=O)C[C@@H](N)CCCN)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CNC(=O)[C@@H]1[C@@H]1NC(N)=NCC1 VCOPTHOUUNAYKQ-WBTCAYNUSA-N 0.000 description 1
- CUKWUWBLQQDQAC-VEQWQPCFSA-N (3s)-3-amino-4-[[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2s,3s)-1-[[(2s)-1-[(2s)-2-[[(1s)-1-carboxyethyl]carbamoyl]pyrrolidin-1-yl]-3-(1h-imidazol-5-yl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-methyl-1-ox Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C1=CC=C(O)C=C1 CUKWUWBLQQDQAC-VEQWQPCFSA-N 0.000 description 1
- JWUPWOYNGNTOKD-WCBMZHEXSA-N (3s,5r)-3-(methylamino)-2,3,4,5-tetrahydro-1-benzoxepin-5-ol Chemical compound O[C@@H]1C[C@H](NC)COC2=CC=CC=C21 JWUPWOYNGNTOKD-WCBMZHEXSA-N 0.000 description 1
- CVEKPEDQESUNSP-NVNXTCNLSA-N (3z)-3-(2-methoxyxanthen-9-ylidene)-n,n-dimethylpropan-1-amine Chemical compound C1=CC=C2\C(=C\CCN(C)C)C3=CC(OC)=CC=C3OC2=C1 CVEKPEDQESUNSP-NVNXTCNLSA-N 0.000 description 1
- UJAOSPFULOFZRR-UHFFFAOYSA-N (4-acetamidophenyl) acetate Chemical compound CC(=O)NC1=CC=C(OC(C)=O)C=C1 UJAOSPFULOFZRR-UHFFFAOYSA-N 0.000 description 1
- NIVDRJXCJCEFDD-UHFFFAOYSA-N (4-chlorophenyl) 2-(4-chlorophenoxy)-2-methylpropanoate Chemical compound C=1C=C(Cl)C=CC=1OC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 NIVDRJXCJCEFDD-UHFFFAOYSA-N 0.000 description 1
- DITYEPYMBCHKLF-UHFFFAOYSA-N (4-chlorophenyl)-(4,5-dihydro-1h-imidazol-2-yl)-pyridin-2-ylmethanol Chemical compound C=1C=C(Cl)C=CC=1C(C=1N=CC=CC=1)(O)C1=NCCN1 DITYEPYMBCHKLF-UHFFFAOYSA-N 0.000 description 1
- XMUZRUCADGTCPX-UHFFFAOYSA-N (4-fluorophenyl)-[1-[3-[2-(trifluoromethyl)phenothiazin-10-yl]propyl]piperidin-4-yl]methanone Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCCN2C3=CC(=CC=C3SC3=CC=CC=C32)C(F)(F)F)CC1 XMUZRUCADGTCPX-UHFFFAOYSA-N 0.000 description 1
- PPQZABOURJVKNI-UHFFFAOYSA-N (4-fluorophenyl)-[4-(4-fluorophenyl)-4-hydroxy-1-methylpiperidin-3-yl]methanone Chemical compound C1N(C)CCC(O)(C=2C=CC(F)=CC=2)C1C(=O)C1=CC=C(F)C=C1 PPQZABOURJVKNI-UHFFFAOYSA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- JMPUQJQXECRLKY-IKADHJCRSA-N (4r,4ar,5s,7ar,12bs)-3-(cyclopropylmethyl)-5-ethyl-9-methoxy-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline-7-one Chemical compound C([C@@]12[C@@H]3[C@H]4CC=5C2=C(C(=CC=5)OC)O[C@H]1C(=O)C[C@@H]3CC)CN4CC1CC1 JMPUQJQXECRLKY-IKADHJCRSA-N 0.000 description 1
- SGKRLCUYIXIAHR-AKNGSSGZSA-N (4s,4ar,5s,5ar,6r,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1=CC=C2[C@H](C)[C@@H]([C@H](O)[C@@H]3[C@](C(O)=C(C(N)=O)C(=O)[C@H]3N(C)C)(O)C3=O)C3=C(O)C2=C1O SGKRLCUYIXIAHR-AKNGSSGZSA-N 0.000 description 1
- RMVMLZHPWMTQGK-SOUFLCLCSA-N (4s,4as,5as,6s,12ar)-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1([C@H]2O)=CC=CC(O)=C1C(O)=C1[C@@H]2C[C@H]2[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]2(O)C1=O RMVMLZHPWMTQGK-SOUFLCLCSA-N 0.000 description 1
- GUXHBMASAHGULD-SEYHBJAFSA-N (4s,4as,5as,6s,12ar)-7-chloro-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1([C@H]2O)=C(Cl)C=CC(O)=C1C(O)=C1[C@@H]2C[C@H]2[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]2(O)C1=O GUXHBMASAHGULD-SEYHBJAFSA-N 0.000 description 1
- FAMUIRDLAWWMCQ-AQFAATAFSA-N (4s,4as,5as,6s,12ar)-n-[[4-[n-(diaminomethylidene)carbamimidoyl]piperazin-1-yl]methyl]-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound OC([C@@]1(O)C(=O)C=2[C@@H]([C@](C3=CC=CC(O)=C3C=2O)(C)O)C[C@H]1[C@@H](C1=O)N(C)C)=C1C(=O)NCN1CCN(C(=N)N=C(N)N)CC1 FAMUIRDLAWWMCQ-AQFAATAFSA-N 0.000 description 1
- IJTPLVAAROHGGB-UHFFFAOYSA-N (5,7-dibromo-2-methylquinolin-8-yl) benzoate Chemical compound C12=NC(C)=CC=C2C(Br)=CC(Br)=C1OC(=O)C1=CC=CC=C1 IJTPLVAAROHGGB-UHFFFAOYSA-N 0.000 description 1
- KEZAYQGUTKCBLO-UHFFFAOYSA-N (5,7-dichloro-1,3-benzoxazol-2-yl)methylsulfanyl-diethoxy-sulfanylidene-$l^{5}-phosphane Chemical compound ClC1=CC(Cl)=C2OC(CSP(=S)(OCC)OCC)=NC2=C1 KEZAYQGUTKCBLO-UHFFFAOYSA-N 0.000 description 1
- AMMYEZPDRKXESR-XHSUIHOPSA-N (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl (6r,7r)-7-[[(2r)-2-[(2s)-2-aminopropanoyl]oxy-2-phenylacetyl]amino]-3-[(5-methyl-1,3,4-thiadiazol-2-yl)sulfanylmethyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate Chemical compound S([C@@H]1[C@@H](C(N1C=1C(=O)OCC2=C(OC(=O)O2)C)=O)NC(=O)[C@H](OC(=O)[C@@H](N)C)C=2C=CC=CC=2)CC=1CSC1=NN=C(C)S1 AMMYEZPDRKXESR-XHSUIHOPSA-N 0.000 description 1
- UJEPHPADGSWWRM-LDYMZIIASA-N (5R,6S)-5-methyl-6-phenylmorpholin-3-one Chemical compound C[C@H]1NC(=O)CO[C@H]1C1=CC=CC=C1 UJEPHPADGSWWRM-LDYMZIIASA-N 0.000 description 1
- DKRRNPBLGINDKV-PHCWAFMCSA-N (5R,8S,9S,10R,13S,14S)-3-fluoro-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-4-ol Chemical compound FC1C([C@@H]2CC[C@@H]3[C@H](CC[C@@]4(CCC[C@H]43)C)[C@]2(CC1)C)O DKRRNPBLGINDKV-PHCWAFMCSA-N 0.000 description 1
- VVLJQSJNPKNTAT-SNVBAGLBSA-N (5S)-5-phenyl-2,3,5,6-tetrahydro-1H-imidazo[1,2-a]imidazole Chemical compound C1([C@@H]2N3CCNC3=NC2)=CC=CC=C1 VVLJQSJNPKNTAT-SNVBAGLBSA-N 0.000 description 1
- MMQXRUYUBYWTDP-KEIGCJKLSA-N (5s,6r,7r)-3-(acetyloxymethyl)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-5,8-dioxo-5$l^{4}-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@@H]1C(N2[C@@H]1[S@](CC(COC(C)=O)=C2C(O)=O)=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 MMQXRUYUBYWTDP-KEIGCJKLSA-N 0.000 description 1
- DIPBYRMTSXMCIM-MOZMXQSSSA-N (6ar,9r,10ar)-n-[(e,2r,3s)-1,3-dihydroxyoctadec-4-en-2-yl]-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline-9-carboxamide Chemical compound C1=CC([C@H]2C[C@H](CN(C)[C@@H]2C2)C(=O)N[C@H](CO)[C@@H](O)/C=C/CCCCCCCCCCCCC)=C3C2=CNC3=C1 DIPBYRMTSXMCIM-MOZMXQSSSA-N 0.000 description 1
- YHEIHLVIKSTGJE-YXJHDRRASA-N (6ar,9s,10ar)-9-(diethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(CC)CC)=C3C2=CNC3=C1 YHEIHLVIKSTGJE-YXJHDRRASA-N 0.000 description 1
- VUEGYUOUAAVYAS-JGGQBBKZSA-N (6ar,9s,10ar)-9-(dimethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CNC3=C1 VUEGYUOUAAVYAS-JGGQBBKZSA-N 0.000 description 1
- YBHZVPYSEUQIII-DYJQDLSISA-N (6r,7r)-3-(acetyloxymethyl)-7-[[(2z)-2-(furan-2-yl)-2-methoxyiminoacetyl]amino]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@@H]1C(N2C(=C(COC(C)=O)CS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CC=CO1 YBHZVPYSEUQIII-DYJQDLSISA-N 0.000 description 1
- OCLRGULJISNUQS-OXQOHEQNSA-N (6r,7r)-3-(acetyloxymethyl)-7-[[3-(2-chlorophenyl)-5-methyl-1,2-oxazole-4-carbonyl]amino]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC(=O)C)C(O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1Cl OCLRGULJISNUQS-OXQOHEQNSA-N 0.000 description 1
- QFTZCQVZVRVDTD-DNVCBOLYSA-N (6r,7r)-3-methyl-8-oxo-7-[[2-[4-(1,4,5,6-tetrahydropyrimidin-2-yl)phenyl]acetyl]amino]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)C)C(O)=O)C(=O)CC(C=C1)=CC=C1C1=NCCCN1 QFTZCQVZVRVDTD-DNVCBOLYSA-N 0.000 description 1
- XSPUSVIQHBDITA-KXDGEKGBSA-N (6r,7r)-7-[[(2e)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-3-[(5-methyltetrazol-2-yl)methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)/C(=N/OC)C=2N=C(N)SC=2)CC=1CN1N=NC(C)=N1 XSPUSVIQHBDITA-KXDGEKGBSA-N 0.000 description 1
- RULITNAIJFZYLO-UEKVPHQBSA-N (6r,7r)-7-[[(2r)-2-amino-2-(3-chloro-4-hydroxyphenyl)acetyl]amino]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=C(O)C(Cl)=C1 RULITNAIJFZYLO-UEKVPHQBSA-N 0.000 description 1
- SBUCDZYLTRYMFG-PBFPGSCMSA-N (6r,7r)-7-[[(2r)-2-amino-2-(4-hydroxyphenyl)acetyl]amino]-3-[(5-methyl-1,3,4-thiadiazol-2-yl)sulfanylmethyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound S1C(C)=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)[C@H](N)C=3C=CC(O)=CC=3)[C@H]2SC1 SBUCDZYLTRYMFG-PBFPGSCMSA-N 0.000 description 1
- VDFFPBOAOLQAJV-SUYBPPKGSA-N (6r,7r)-7-[[(2r)-2-hydroxy-2-phenylacetyl]amino]-3-[(5-methyl-1,3,4-thiadiazol-2-yl)sulfanylmethyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound S1C(C)=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)[C@H](O)C=3C=CC=CC=3)[C@H]2SC1 VDFFPBOAOLQAJV-SUYBPPKGSA-N 0.000 description 1
- WYKASFRWOOMFNP-ZISBYHFMSA-N (6r,7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-(2-carboxypropan-2-yloxyimino)acetyl]amino]-3-[(1-methylpyridin-1-ium-4-yl)sulfanylmethyl]-5,8-dioxo-5$l^{4}-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid;chloride Chemical compound [Cl-].C1=C[N+](C)=CC=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)C(=N/OC(C)(C)C(O)=O)\C=3N=C(N)SC=3)[C@H]2S(=O)C1 WYKASFRWOOMFNP-ZISBYHFMSA-N 0.000 description 1
- YWKJNRNSJKEFMK-PQFQYKRASA-N (6r,7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-8-oxo-3-(5,6,7,8-tetrahydroquinolin-1-ium-1-ylmethyl)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate Chemical compound N([C@@H]1C(N2C(=C(C[N+]=3C=4CCCCC=4C=CC=3)CS[C@@H]21)C([O-])=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 YWKJNRNSJKEFMK-PQFQYKRASA-N 0.000 description 1
- CUCFRCCRYQDMNM-PXIRVSTKSA-N (6r,7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-5-yl)-2-(2-oxopyrrolidin-3-yl)oxyiminoacetyl]amino]-8-oxo-3-(pyridin-1-ium-1-ylmethyl)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate Chemical compound S1C(N)=NC=C1C(\C(=O)N[C@@H]1C(N2C(=C(C[N+]=3C=CC=CC=3)CS[C@@H]21)C([O-])=O)=O)=N\OC1C(=O)NCC1 CUCFRCCRYQDMNM-PXIRVSTKSA-N 0.000 description 1
- UQWYUAURRDNBKR-BXUZGUMPSA-N (6r,7r)-8-oxo-7-[(2-thiophen-2-ylacetyl)amino]-3-(1h-1,2,4-triazol-5-ylsulfanylmethyl)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound O=C([C@@H](NC(=O)CC=1SC=CC=1)[C@H]1SC2)N1C(C(=O)O)=C2CSC1=NC=NN1 UQWYUAURRDNBKR-BXUZGUMPSA-N 0.000 description 1
- RNSISAJHBSCDGC-SCUQKFFVSA-N (6s,8r,9s,10r,13s,14s,17r)-17-acetyl-6-chloro-17-hydroxy-10,13-dimethyl-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-3-one Chemical compound C1([C@@H](Cl)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@@](C(=O)C)(O)[C@@]2(C)CC1 RNSISAJHBSCDGC-SCUQKFFVSA-N 0.000 description 1
- VDNZZIYSCXESNI-ILSZZQPISA-N (6s,8s,9s,10r,11s,13s,14s,17s)-17-acetyl-11-hydroxy-6,10,13-trimethyl-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-3-one Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@H](C(C)=O)CC[C@H]21 VDNZZIYSCXESNI-ILSZZQPISA-N 0.000 description 1
- JTVLYHXMPUSZIT-VKAVYKQESA-N (6z)-6-[butylamino-(2-chlorophenyl)methylidene]-4-chlorocyclohexa-2,4-dien-1-one Chemical compound C=1C=CC=C(Cl)C=1C(/NCCCC)=C1\C=C(Cl)C=CC1=O JTVLYHXMPUSZIT-VKAVYKQESA-N 0.000 description 1
- XDGZWAFPWNIYGI-SAPRIBHFSA-N (8R,9S,10R,13S,14S)-10,13-dimethyl-1,2,3,6,7,8,9,11,12,14-decahydrocyclopenta[a]phenanthrene-15,16,17-trione Chemical compound [C@@H]12C(C(C([C@@]1(C)CC[C@H]1[C@H]2CCC2=CCCC[C@]12C)=O)=O)=O XDGZWAFPWNIYGI-SAPRIBHFSA-N 0.000 description 1
- YXWAYKWHUNYBMS-QJBNOVHISA-N (8R,9S,10R,13S,14S,17S)-15,17-dihydroxy-10,13,15,16-tetramethyl-2,6,7,8,9,11,12,14,16,17-decahydro-1H-cyclopenta[a]phenanthren-3-one Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@]4(C)[C@@H](O)C(C)C(O)(C)[C@H]4[C@@H]3CCC2=C1 YXWAYKWHUNYBMS-QJBNOVHISA-N 0.000 description 1
- HSYWFJBHXIUUCZ-XGXHKTLJSA-N (8r,9s,10r,13s,14s,17r)-17-ethynyl-13-methyl-2,3,4,7,8,9,10,11,12,14,15,16-dodecahydro-1h-cyclopenta[a]phenanthren-17-ol Chemical compound C1CCC[C@@H]2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CC=C21 HSYWFJBHXIUUCZ-XGXHKTLJSA-N 0.000 description 1
- XMPHGFYWQPUCAG-XMUHMHRVSA-N (8r,9s,10r,13s,14s,17s)-4,17-dihydroxy-10,13,17-trimethyl-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-3-one Chemical compound C1CC2=C(O)C(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)CC2 XMPHGFYWQPUCAG-XMUHMHRVSA-N 0.000 description 1
- JVLHMVXGPLKLFV-AJUWMHLFSA-N (8r,9s,13s,14s,17s)-13-methyl-17-(2,2,2-trichloro-1-hydroxyethoxy)-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-3-ol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)OC(O)C(Cl)(Cl)Cl)[C@@H]4[C@@H]3CCC2=C1 JVLHMVXGPLKLFV-AJUWMHLFSA-N 0.000 description 1
- KEOBKPHJNAILCW-FUMNGEBKSA-N (8s,13s,14s,17s)-17-(2-chloroethynyl)-17-hydroxy-13-methyl-1,2,6,7,8,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-3-one Chemical compound C1CC(=O)C=C2CC[C@@H]([C@H]3[C@@](C)([C@](CC3)(O)C#CCl)CC3)C3=C21 KEOBKPHJNAILCW-FUMNGEBKSA-N 0.000 description 1
- FXBVINCSIJOIGL-ODTHGOCZSA-N (8s,9r,10s,11s,13s,14s,16r,17s)-9-fluoro-11-hydroxy-17-(2-hydroxyacetyl)-10,13,16,17-tetramethyl-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-3-one Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(C)[C@@]1(C)C[C@@H]2O FXBVINCSIJOIGL-ODTHGOCZSA-N 0.000 description 1
- IKGBPSZWCRRUQS-DTAAKRQUSA-N (8s,9r,10s,11s,13s,14s,16s,17r)-17-acetyl-9-fluoro-11,17-dihydroxy-10,13,16-trimethyl-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-3-one Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(C)=O)(O)[C@@]1(C)C[C@@H]2O IKGBPSZWCRRUQS-DTAAKRQUSA-N 0.000 description 1
- CZJXBZPJABCCRQ-BULBTXNYSA-N (8s,9r,10s,11s,13s,14s,17r)-9,11-dichloro-17-hydroxy-17-(2-hydroxyacetyl)-10,13-dimethyl-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-3-one Chemical compound O=C1C=C[C@]2(C)[C@@]3(Cl)[C@@H](Cl)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 CZJXBZPJABCCRQ-BULBTXNYSA-N 0.000 description 1
- KQZSMOGWYFPKCH-UJPCIWJBSA-N (8s,9s,10r,11s,13s,14s,17r)-17-acetyl-11,17-dihydroxy-10,13-dimethyl-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-3-one Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)C[C@@H]2O KQZSMOGWYFPKCH-UJPCIWJBSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- 229930182837 (R)-adrenaline Natural products 0.000 description 1
- XUCIJNAGGSZNQT-JHSLDZJXSA-N (R)-amygdalin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@H](O[C@@H](C#N)C=2C=CC=CC=2)O1 XUCIJNAGGSZNQT-JHSLDZJXSA-N 0.000 description 1
- PHIQHXFUZVPYII-ZCFIWIBFSA-N (R)-carnitine Chemical compound C[N+](C)(C)C[C@H](O)CC([O-])=O PHIQHXFUZVPYII-ZCFIWIBFSA-N 0.000 description 1
- WSEQXVZVJXJVFP-HXUWFJFHSA-N (R)-citalopram Chemical compound C1([C@@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-HXUWFJFHSA-N 0.000 description 1
- XFDJYSQDBULQSI-QFIPXVFZSA-N (R)-doxapram Chemical compound C([C@H]1CN(C(C1(C=1C=CC=CC=1)C=1C=CC=CC=1)=O)CC)CN1CCOCC1 XFDJYSQDBULQSI-QFIPXVFZSA-N 0.000 description 1
- MHFRGQHAERHWKZ-HHHXNRCGSA-N (R)-edelfosine Chemical compound CCCCCCCCCCCCCCCCCCOC[C@@H](OC)COP([O-])(=O)OCC[N+](C)(C)C MHFRGQHAERHWKZ-HHHXNRCGSA-N 0.000 description 1
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 1
- SGUAFYQXFOLMHL-ACJLOTCBSA-N (R,R)-labetalol Chemical compound C([C@@H](C)NC[C@H](O)C=1C=C(C(O)=CC=1)C(N)=O)CC1=CC=CC=C1 SGUAFYQXFOLMHL-ACJLOTCBSA-N 0.000 description 1
- MXOAEAUPQDYUQM-QMMMGPOBSA-N (S)-chlorphenesin Chemical compound OC[C@H](O)COC1=CC=C(Cl)C=C1 MXOAEAUPQDYUQM-QMMMGPOBSA-N 0.000 description 1
- SYTBZMRGLBWNTM-JTQLQIEISA-N (S)-flurbiprofen Chemical compound FC1=CC([C@@H](C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-JTQLQIEISA-N 0.000 description 1
- WSPOMRSOLSGNFJ-AUWJEWJLSA-N (Z)-chlorprothixene Chemical compound C1=C(Cl)C=C2C(=C/CCN(C)C)\C3=CC=CC=C3SC2=C1 WSPOMRSOLSGNFJ-AUWJEWJLSA-N 0.000 description 1
- WSJULBMCKQTTIG-OWOJBTEDSA-N (e)-1,1,1,2,3,4,4,4-octafluorobut-2-ene Chemical compound FC(F)(F)C(/F)=C(\F)C(F)(F)F WSJULBMCKQTTIG-OWOJBTEDSA-N 0.000 description 1
- HIPIFFZGPOCRPX-ZZXKWVIFSA-N (e)-1-(2,4-dimethoxyphenyl)-3-pyridin-4-ylprop-2-en-1-one Chemical compound COC1=CC(OC)=CC=C1C(=O)\C=C\C1=CC=NC=C1 HIPIFFZGPOCRPX-ZZXKWVIFSA-N 0.000 description 1
- AGNGYMCLFWQVGX-AGFFZDDWSA-N (e)-1-[(2s)-2-amino-2-carboxyethoxy]-2-diazonioethenolate Chemical compound OC(=O)[C@@H](N)CO\C([O-])=C\[N+]#N AGNGYMCLFWQVGX-AGFFZDDWSA-N 0.000 description 1
- SYFDPNLESAYNCB-VMPITWQZSA-N (e)-1-[4-[3-[(8-acetyl-2,3-dihydro-1,4-benzodioxin-5-yl)oxy]-2-hydroxypropyl]piperazin-1-yl]-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one Chemical compound COC1=C(OC)C(OC)=CC(\C=C\C(=O)N2CCN(CC(O)COC=3C=4OCCOC=4C(C(C)=O)=CC=3)CC2)=C1 SYFDPNLESAYNCB-VMPITWQZSA-N 0.000 description 1
- LRLKZVMLJBNNPE-SNAWJCMRSA-N (e)-3-(3,4,5-trimethoxyphenyl)prop-2-enamide Chemical compound COC1=CC(\C=C\C(N)=O)=CC(OC)=C1OC LRLKZVMLJBNNPE-SNAWJCMRSA-N 0.000 description 1
- NKPCAAMLVDTZOB-ZHACJKMWSA-N (e)-4-[3,4-bis(4-chlorophenyl)butan-2-ylamino]-4-oxobut-2-enoic acid Chemical compound C=1C=C(Cl)C=CC=1C(C(NC(=O)\C=C\C(O)=O)C)CC1=CC=C(Cl)C=C1 NKPCAAMLVDTZOB-ZHACJKMWSA-N 0.000 description 1
- LBOZSXSPRGACHC-NFOZGECASA-L (e)-bis(3-methyl-2-phenylimidazo[1,2-a]pyridin-4-ium-1-yl)diazene;dibromide Chemical compound [Br-].[Br-].C=1C=CC=CC=1C1=C(C)[N+]2=CC=CC=C2N1/N=N/N1C2=CC=CC=[N+]2C(C)=C1C1=CC=CC=C1 LBOZSXSPRGACHC-NFOZGECASA-L 0.000 description 1
- QTKQVDXGCWKEHE-SDNWHVSQSA-N (e)-n-(2-benzhydryloxyethyl)-n-methyl-3-phenylprop-2-en-1-amine Chemical compound C=1C=CC=CC=1/C=C/CN(C)CCOC(C=1C=CC=CC=1)C1=CC=CC=C1 QTKQVDXGCWKEHE-SDNWHVSQSA-N 0.000 description 1
- QCUPYFTWJOZAOB-HWKANZROSA-N (e)-n-carbamoyl-2-ethylbut-2-enamide Chemical compound CC\C(=C/C)C(=O)NC(N)=O QCUPYFTWJOZAOB-HWKANZROSA-N 0.000 description 1
- NCOOUEIQXVWKTO-QPJJXVBHSA-N (e)-n-cyclopropyl-3-(3-fluorophenyl)prop-2-enamide Chemical compound FC1=CC=CC(\C=C\C(=O)NC2CC2)=C1 NCOOUEIQXVWKTO-QPJJXVBHSA-N 0.000 description 1
- WDMFHQNUSVLQMS-XFYQWYMDSA-N (e,5z)-5-(3,3-dimethyl-2-bicyclo[2.2.1]heptanylidene)pent-3-en-2-one Chemical compound C1CC2C(C)(C)C(=C/C=C/C(=O)C)\C1C2 WDMFHQNUSVLQMS-XFYQWYMDSA-N 0.000 description 1
- CADPBCPVJBOUEG-HMMYKYKNSA-N (ne)-n-(9,10-dimethoxy-1,2,3,4,4a,6,7,11b,12,13a-decahydroisoquinolino[2,1-a]quinolin-13-ylidene)hydroxylamine Chemical compound C12CCCCC2\C(=N\O)CC2N1CCC1=C2C=C(OC)C(OC)=C1 CADPBCPVJBOUEG-HMMYKYKNSA-N 0.000 description 1
- PNWRDBWQKOXCMK-JQIJEIRASA-N (ne)-n-[1-[1-[2-(dimethylamino)ethyl]indol-3-yl]propylidene]hydroxylamine Chemical compound C1=CC=C2C(C(=N/O)/CC)=CN(CCN(C)C)C2=C1 PNWRDBWQKOXCMK-JQIJEIRASA-N 0.000 description 1
- CAFOIGUDKPQBIO-BYIOMEFUSA-N (r)-[(2s,4s,5r)-5-ethyl-1-azabicyclo[2.2.2]octan-2-yl]-[6-(3-methylbutoxy)quinolin-4-yl]methanol Chemical compound C1=C(OCCC(C)C)C=C2C([C@@H](O)[C@@H]3C[C@@H]4CCN3C[C@@H]4CC)=CC=NC2=C1 CAFOIGUDKPQBIO-BYIOMEFUSA-N 0.000 description 1
- KDVWQPQZDKHFOC-PTRXCMEYSA-N (z)-7-[(1r,2r,3r,5r)-5-fluoro-3-hydroxy-2-[(e,3r)-3-hydroxy-4-phenoxybut-1-enyl]cyclopentyl]hept-5-enoic acid Chemical compound C([C@H](O)\C=C\[C@@H]1[C@H]([C@H](F)C[C@H]1O)C\C=C/CCCC(O)=O)OC1=CC=CC=C1 KDVWQPQZDKHFOC-PTRXCMEYSA-N 0.000 description 1
- BPALXSWPHQNCAA-RLJNYRALSA-N (z)-7-[(1r,2r,3r,5s)-3,5-dihydroxy-2-[(e)-2-[2-(phenoxymethyl)-1,3-dioxolan-2-yl]ethenyl]cyclopentyl]hept-5-enoic acid Chemical compound OC(=O)CCC\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\C1(COC=2C=CC=CC=2)OCCO1 BPALXSWPHQNCAA-RLJNYRALSA-N 0.000 description 1
- UJFDLBWYYFZXDS-LEENOTDESA-N (z)-7-[(1s,2s,3s)-2-[(e,3s)-5-ethoxy-3-hydroxy-4,4-dimethylpent-1-enyl]-3-hydroxy-5-oxocyclopentyl]hept-5-enoic acid Chemical compound CCOCC(C)(C)[C@@H](O)\C=C\[C@@H]1[C@@H](O)CC(=O)[C@H]1C\C=C/CCCC(O)=O UJFDLBWYYFZXDS-LEENOTDESA-N 0.000 description 1
- COQIQRBKEGPRSG-UHFFFAOYSA-N 1,1,1,2,3,3,3-heptafluoro-2-(trifluoromethyl)propane Chemical compound FC(F)(F)C(F)(C(F)(F)F)C(F)(F)F COQIQRBKEGPRSG-UHFFFAOYSA-N 0.000 description 1
- MEKBIVQWPIRQNE-UHFFFAOYSA-N 1,1,1-trifluoropropan-2-yl 2-cyanoprop-2-enoate Chemical compound FC(F)(F)C(C)OC(=O)C(=C)C#N MEKBIVQWPIRQNE-UHFFFAOYSA-N 0.000 description 1
- FNVLGCVAWPSVSK-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6,7,7-tetradecafluorocycloheptane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F FNVLGCVAWPSVSK-UHFFFAOYSA-N 0.000 description 1
- RKIMETXDACNTIE-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6-dodecafluorocyclohexane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F RKIMETXDACNTIE-UHFFFAOYSA-N 0.000 description 1
- QIROQPWSJUXOJC-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6-undecafluoro-6-(trifluoromethyl)cyclohexane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F QIROQPWSJUXOJC-UHFFFAOYSA-N 0.000 description 1
- PWMJXZJISGDARB-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5-decafluorocyclopentane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F PWMJXZJISGDARB-UHFFFAOYSA-N 0.000 description 1
- BCNXQFASJTYKDJ-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5-nonafluoro-5-(trifluoromethyl)cyclopentane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F BCNXQFASJTYKDJ-UHFFFAOYSA-N 0.000 description 1
- CIWUYWQUYMZILR-UHFFFAOYSA-N 1,1,2,2,3,3,4,4-octafluoro-5,5-bis(trifluoromethyl)cyclopentane Chemical class FC(F)(F)C1(C(F)(F)F)C(F)(F)C(F)(F)C(F)(F)C1(F)F CIWUYWQUYMZILR-UHFFFAOYSA-N 0.000 description 1
- ZVXOHSHODRJTCP-UHFFFAOYSA-N 1,1,2,2,3,3,4-heptafluoro-4-(trifluoromethyl)cyclobutane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C1(F)F ZVXOHSHODRJTCP-UHFFFAOYSA-N 0.000 description 1
- TXGPGHBYAPBDAG-UHFFFAOYSA-N 1,1,2,2,3,3-hexafluoro-4,4-bis(trifluoromethyl)cyclobutane Chemical class FC(F)(F)C1(C(F)(F)F)C(F)(F)C(F)(F)C1(F)F TXGPGHBYAPBDAG-UHFFFAOYSA-N 0.000 description 1
- YUFJLVUCHXMKKM-UHFFFAOYSA-N 1,1,2,2,3-pentafluoro-3,4,4-tris(trifluoromethyl)cyclobutane Chemical class FC(F)(F)C1(F)C(F)(F)C(F)(F)C1(C(F)(F)F)C(F)(F)F YUFJLVUCHXMKKM-UHFFFAOYSA-N 0.000 description 1
- LGPPATCNSOSOQH-UHFFFAOYSA-N 1,1,2,3,4,4-hexafluorobuta-1,3-diene Chemical compound FC(F)=C(F)C(F)=C(F)F LGPPATCNSOSOQH-UHFFFAOYSA-N 0.000 description 1
- NPNPZTNLOVBDOC-UHFFFAOYSA-N 1,1-difluoroethane Chemical compound CC(F)F NPNPZTNLOVBDOC-UHFFFAOYSA-N 0.000 description 1
- IBMCQJYLPXUOKM-UHFFFAOYSA-N 1,2,2,6,6-pentamethyl-3h-pyridine Chemical compound CN1C(C)(C)CC=CC1(C)C IBMCQJYLPXUOKM-UHFFFAOYSA-N 0.000 description 1
- ZTXDHEQQZVFGPK-UHFFFAOYSA-N 1,2,4-tris(oxiran-2-ylmethyl)-1,2,4-triazolidine-3,5-dione Chemical compound C1OC1CN1C(=O)N(CC2OC2)C(=O)N1CC1CO1 ZTXDHEQQZVFGPK-UHFFFAOYSA-N 0.000 description 1
- XKZKYMVAWQDWMV-UHFFFAOYSA-N 1,2-bis(aziridin-1-yl)butane-1,1-diol Chemical compound C1CN1C(O)(O)C(CC)N1CC1 XKZKYMVAWQDWMV-UHFFFAOYSA-N 0.000 description 1
- CITHEXJVPOWHKC-UUWRZZSWSA-N 1,2-di-O-myristoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCC CITHEXJVPOWHKC-UUWRZZSWSA-N 0.000 description 1
- KILNVBDSWZSGLL-KXQOOQHDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCC KILNVBDSWZSGLL-KXQOOQHDSA-N 0.000 description 1
- SLKDGVPOSSLUAI-PGUFJCEWSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine zwitterion Chemical group CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OCCN)OC(=O)CCCCCCCCCCCCCCC SLKDGVPOSSLUAI-PGUFJCEWSA-N 0.000 description 1
- MTZUIIAIAKMWLI-UHFFFAOYSA-N 1,2-diisocyanatobenzene Chemical compound O=C=NC1=CC=CC=C1N=C=O MTZUIIAIAKMWLI-UHFFFAOYSA-N 0.000 description 1
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 description 1
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 1
- PRXHANOJNVGVGC-UHFFFAOYSA-N 1,3-bis(4-ethoxyphenyl)thiourea Chemical compound C1=CC(OCC)=CC=C1NC(=S)NC1=CC=C(OCC)C=C1 PRXHANOJNVGVGC-UHFFFAOYSA-N 0.000 description 1
- YDDUMTOHNYZQPO-UHFFFAOYSA-N 1,3-bis{[(2E)-3-(3,4-dihydroxyphenyl)prop-2-enoyl]oxy}-4,5-dihydroxycyclohexanecarboxylic acid Natural products OC1C(O)CC(C(O)=O)(OC(=O)C=CC=2C=C(O)C(O)=CC=2)CC1OC(=O)C=CC1=CC=C(O)C(O)=C1 YDDUMTOHNYZQPO-UHFFFAOYSA-N 0.000 description 1
- ABSDZQDZDHJMPX-UHFFFAOYSA-N 1,3-dibutoxypropan-2-ol Chemical compound CCCCOCC(O)COCCCC ABSDZQDZDHJMPX-UHFFFAOYSA-N 0.000 description 1
- HJPRDDKCXVCFOH-UHFFFAOYSA-N 1,3-dibutyl-7-(2-oxopropyl)purine-2,6-dione Chemical compound O=C1N(CCCC)C(=O)N(CCCC)C2=C1N(CC(C)=O)C=N2 HJPRDDKCXVCFOH-UHFFFAOYSA-N 0.000 description 1
- MVXGSLGVWBVZCA-UHFFFAOYSA-N 1,3-dimethyl-7-[2-(1-phenylpropan-2-ylamino)ethyl]purine-2,6-dione;hydrochloride Chemical compound Cl.C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCNC(C)CC1=CC=CC=C1 MVXGSLGVWBVZCA-UHFFFAOYSA-N 0.000 description 1
- PVKUHCBHOUMGJN-UHFFFAOYSA-N 1,3-dimethyl-7h-purine-2,6-dione;3-(2-methoxyphenoxy)propane-1,2-diol Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2.COC1=CC=CC=C1OCC(O)CO PVKUHCBHOUMGJN-UHFFFAOYSA-N 0.000 description 1
- HNSDLXPSAYFUHK-UHFFFAOYSA-N 1,4-bis(2-ethylhexyl) sulfosuccinate Chemical compound CCCCC(CC)COC(=O)CC(S(O)(=O)=O)C(=O)OCC(CC)CCCC HNSDLXPSAYFUHK-UHFFFAOYSA-N 0.000 description 1
- RKDVKSZUMVYZHH-UHFFFAOYSA-N 1,4-dioxane-2,5-dione Chemical group O=C1COC(=O)CO1 RKDVKSZUMVYZHH-UHFFFAOYSA-N 0.000 description 1
- VILFTWLXLYIEMV-UHFFFAOYSA-N 1,5-difluoro-2,4-dinitrobenzene Chemical compound [O-][N+](=O)C1=CC([N+]([O-])=O)=C(F)C=C1F VILFTWLXLYIEMV-UHFFFAOYSA-N 0.000 description 1
- JPKRUWRMHGVJCV-UHFFFAOYSA-N 1,5-dimethyl-4-[(2-methyl-5-oxo-1-phenyl-4-propan-2-ylpyrazol-3-yl)methylamino]-2-phenylpyrazol-3-one Chemical compound CN1N(C=2C=CC=CC=2)C(=O)C(C(C)C)=C1CNC(C1=O)=C(C)N(C)N1C1=CC=CC=C1 JPKRUWRMHGVJCV-UHFFFAOYSA-N 0.000 description 1
- WEEGYLXZBRQIMU-UHFFFAOYSA-N 1,8-cineole Natural products C1CC2CCC1(C)OC2(C)C WEEGYLXZBRQIMU-UHFFFAOYSA-N 0.000 description 1
- VPBLOJFGPORKQA-UHFFFAOYSA-N 1-(1-adamantyl)azetidine-2-carboxylic acid Chemical compound OC(=O)C1CCN1C1(C2)CC(C3)CC2CC3C1 VPBLOJFGPORKQA-UHFFFAOYSA-N 0.000 description 1
- MIKZNWZBJNZVLB-UHFFFAOYSA-N 1-(1-aminopentylidene)-3-(4-chlorophenyl)urea;dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O.CCCC\C(N)=N\C(=O)NC1=CC=C(Cl)C=C1 MIKZNWZBJNZVLB-UHFFFAOYSA-N 0.000 description 1
- UOQIGMZTLWHOOA-UHFFFAOYSA-N 1-(1-ethynylcyclohexyl)oxy-3-[4-(4-fluorophenyl)piperazin-1-yl]propan-2-ol Chemical compound C1CN(C=2C=CC(F)=CC=2)CCN1CC(O)COC1(C#C)CCCCC1 UOQIGMZTLWHOOA-UHFFFAOYSA-N 0.000 description 1
- HYMMMXNCSPCFPW-UHFFFAOYSA-N 1-(1-methylbenzimidazol-2-yl)ethyl n-[4-[1-(1-methylbenzimidazol-2-yl)ethylsulfanylcarbothioylamino]phenyl]carbamodithioate Chemical compound C1=CC=C2N(C)C(C(SC(=S)NC=3C=CC(NC(=S)SC(C)C=4N(C5=CC=CC=C5N=4)C)=CC=3)C)=NC2=C1 HYMMMXNCSPCFPW-UHFFFAOYSA-N 0.000 description 1
- WQFUGXLJDDEHRM-UHFFFAOYSA-N 1-(2,4-dimethoxyphenyl)-1-phenylethanol Chemical compound COC1=CC(OC)=CC=C1C(C)(O)C1=CC=CC=C1 WQFUGXLJDDEHRM-UHFFFAOYSA-N 0.000 description 1
- YJZJEQBSODVMTH-UHFFFAOYSA-N 1-(2-chloroethyl)-3-(2-hydroxyethyl)-1-nitrosourea Chemical compound OCCNC(=O)N(N=O)CCCl YJZJEQBSODVMTH-UHFFFAOYSA-N 0.000 description 1
- XWPCYYOZOJKYKQ-UHFFFAOYSA-N 1-(2-chloroethyl)-3-[2-[2-[[2-chloroethyl(nitroso)carbamoyl]amino]ethyldisulfanyl]ethyl]-1-nitrosourea Chemical compound ClCCN(N=O)C(=O)NCCSSCCNC(=O)N(N=O)CCCl XWPCYYOZOJKYKQ-UHFFFAOYSA-N 0.000 description 1
- ZZKWNLZUYAGVOT-UHFFFAOYSA-N 1-(2-chlorophenothiazin-10-yl)-3-(diethylamino)propan-1-one Chemical compound C1=C(Cl)C=C2N(C(=O)CCN(CC)CC)C3=CC=CC=C3SC2=C1 ZZKWNLZUYAGVOT-UHFFFAOYSA-N 0.000 description 1
- SSMSBSWKLKKXGG-UHFFFAOYSA-N 1-(2-chlorophenyl)-2-isopropylaminoethanol Chemical compound CC(C)NCC(O)C1=CC=CC=C1Cl SSMSBSWKLKKXGG-UHFFFAOYSA-N 0.000 description 1
- ITNSQDCAZVVFMS-UHFFFAOYSA-N 1-(2-methoxyphenyl)-4-(3-methoxypropyl)piperazine Chemical compound C1CN(CCCOC)CCN1C1=CC=CC=C1OC ITNSQDCAZVVFMS-UHFFFAOYSA-N 0.000 description 1
- KJBSVTAYVZKMDM-UHFFFAOYSA-N 1-(2-nitrophenyl)cyclopropane-1-carboxylic acid Chemical compound C=1C=CC=C([N+]([O-])=O)C=1C1(C(=O)O)CC1 KJBSVTAYVZKMDM-UHFFFAOYSA-N 0.000 description 1
- RFZXOOKXYRDPOR-QFFDILLMSA-N 1-(2-piperidin-1-ylethyl)-3-[(e)-[(3z)-3-(2-piperidin-1-ylethylcarbamothioylhydrazinylidene)butan-2-ylidene]amino]thiourea Chemical compound C1CCCCN1CCNC(=S)N/N=C(\C)/C(/C)=N\NC(=S)NCCN1CCCCC1 RFZXOOKXYRDPOR-QFFDILLMSA-N 0.000 description 1
- FEYBPSKVORXMON-UHFFFAOYSA-N 1-(3-chloro-4-cyanophenyl)-3-(diaminomethylidene)urea Chemical compound NC(N)=NC(=O)NC1=CC=C(C#N)C(Cl)=C1 FEYBPSKVORXMON-UHFFFAOYSA-N 0.000 description 1
- DWPQODZAOSWNHB-UHFFFAOYSA-N 1-(3-chlorophenyl)-3-(3-methyl-5-oxo-4H-imidazol-2-yl)urea Chemical compound CN1CC(=O)N=C1NC(=O)NC1=CC=CC(Cl)=C1 DWPQODZAOSWNHB-UHFFFAOYSA-N 0.000 description 1
- KGSABFQIAANNPS-UHFFFAOYSA-N 1-(3-chlorophenyl)-3-(dimethylamino)-1-phenylpropan-2-ol Chemical compound C=1C=CC(Cl)=CC=1C(C(O)CN(C)C)C1=CC=CC=C1 KGSABFQIAANNPS-UHFFFAOYSA-N 0.000 description 1
- OWEGWHBOCFMBLP-UHFFFAOYSA-N 1-(4-chlorophenoxy)-1-(1H-imidazol-1-yl)-3,3-dimethylbutan-2-one Chemical compound C1=CN=CN1C(C(=O)C(C)(C)C)OC1=CC=C(Cl)C=C1 OWEGWHBOCFMBLP-UHFFFAOYSA-N 0.000 description 1
- YYGANUVABKDFDW-UHFFFAOYSA-N 1-(4-chlorophenyl)-3-azabicyclo[3.1.0]hexane-2,4-dione Chemical compound C1=CC(Cl)=CC=C1C1(C(NC2=O)=O)C2C1 YYGANUVABKDFDW-UHFFFAOYSA-N 0.000 description 1
- KVHHQGIIZCJATJ-UHFFFAOYSA-N 1-(4-chlorophenyl)-4-(dimethylamino)-2,3-dimethyl-2-butanol Chemical compound CN(C)CC(C)C(C)(O)CC1=CC=C(Cl)C=C1 KVHHQGIIZCJATJ-UHFFFAOYSA-N 0.000 description 1
- SQUNAWUMZGQQJD-UHFFFAOYSA-N 1-(4-ethylphenyl)-2-methyl-3-(piperidin-1-yl)propan-1-one Chemical compound C1=CC(CC)=CC=C1C(=O)C(C)CN1CCCCC1 SQUNAWUMZGQQJD-UHFFFAOYSA-N 0.000 description 1
- LCZRXFYSMJIDQQ-UHFFFAOYSA-N 1-(4-fluorophenyl)-4-[4-(2-methoxyphenyl)piperazin-1-yl]butan-1-ol Chemical compound COC1=CC=CC=C1N1CCN(CCCC(O)C=2C=CC(F)=CC=2)CC1 LCZRXFYSMJIDQQ-UHFFFAOYSA-N 0.000 description 1
- ICAYNKLSQSKOJZ-UHFFFAOYSA-N 1-(4-fluorophenyl)-4-[4-[(4-fluorophenyl)-hydroxymethyl]piperidin-1-yl]butan-1-one Chemical compound C=1C=C(F)C=CC=1C(O)C(CC1)CCN1CCCC(=O)C1=CC=C(F)C=C1 ICAYNKLSQSKOJZ-UHFFFAOYSA-N 0.000 description 1
- JHBWYQRKOUBPCA-UHFFFAOYSA-N 1-(6-methoxy-1,3-benzothiazol-2-yl)-3-phenylurea Chemical compound S1C2=CC(OC)=CC=C2N=C1NC(=O)NC1=CC=CC=C1 JHBWYQRKOUBPCA-UHFFFAOYSA-N 0.000 description 1
- BAMWZWFFSCYCGQ-UHFFFAOYSA-N 1-(azepan-1-yl)-3-(2,3-dihydro-1h-inden-5-ylsulfonyl)urea Chemical compound C=1C=C2CCCC2=CC=1S(=O)(=O)NC(=O)NN1CCCCCC1 BAMWZWFFSCYCGQ-UHFFFAOYSA-N 0.000 description 1
- VAFNJIFAZJWWNI-UHFFFAOYSA-N 1-(cyclopropylmethyl)-6-methoxy-4-phenylquinazolin-2-one Chemical compound O=C1N=C(C=2C=CC=CC=2)C2=CC(OC)=CC=C2N1CC1CC1 VAFNJIFAZJWWNI-UHFFFAOYSA-N 0.000 description 1
- NCXUNZWLEYGQAH-UHFFFAOYSA-N 1-(dimethylamino)propan-2-ol Chemical compound CC(O)CN(C)C NCXUNZWLEYGQAH-UHFFFAOYSA-N 0.000 description 1
- HMTJIKVWJRSKMY-UXBLZVDNSA-N 1-(furan-2-yl)-3-[2-[[4-[(e)-3-phenylprop-2-enyl]piperazin-1-yl]methyl]benzimidazol-1-yl]propan-1-one Chemical compound C=1C=COC=1C(=O)CCN(C1=CC=CC=C1N=1)C=1CN(CC1)CCN1C\C=C\C1=CC=CC=C1 HMTJIKVWJRSKMY-UXBLZVDNSA-N 0.000 description 1
- SNYUHPPZINRDSG-UHFFFAOYSA-N 1-(oxiran-2-ylmethyl)-4-[1-(oxiran-2-ylmethyl)piperidin-4-yl]piperidine Chemical compound C1CC(C2CCN(CC3OC3)CC2)CCN1CC1CO1 SNYUHPPZINRDSG-UHFFFAOYSA-N 0.000 description 1
- ZJWWWCZRROHPDH-UHFFFAOYSA-N 1-(tert-butylamino)-3-(2,3-dihydro-1,4-benzodioxin-3-ylmethoxy)propan-2-ol Chemical compound C1=CC=C2OC(COCC(O)CNC(C)(C)C)COC2=C1 ZJWWWCZRROHPDH-UHFFFAOYSA-N 0.000 description 1
- UUOJIACWOAYWEZ-UHFFFAOYSA-N 1-(tert-butylamino)-3-[(2-methyl-1H-indol-4-yl)oxy]propan-2-yl benzoate Chemical compound C1=CC=C2NC(C)=CC2=C1OCC(CNC(C)(C)C)OC(=O)C1=CC=CC=C1 UUOJIACWOAYWEZ-UHFFFAOYSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical compound CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- RFCAUADVODFSLZ-UHFFFAOYSA-N 1-Chloro-1,1,2,2,2-pentafluoroethane Chemical compound FC(F)(F)C(F)(F)Cl RFCAUADVODFSLZ-UHFFFAOYSA-N 0.000 description 1
- KOYMTVWMCXDJNV-UHFFFAOYSA-N 1-[(2-methoxy-6-methylpyridin-3-yl)methyl]aziridine-2-carbonitrile Chemical compound COC1=NC(C)=CC=C1CN1C(C#N)C1 KOYMTVWMCXDJNV-UHFFFAOYSA-N 0.000 description 1
- XDTNOYLBDDCJSK-UHFFFAOYSA-N 1-[(3,4-dichlorophenyl)methoxy]-6,6-dimethyl-1,3,5-triazine-2,4-diamine Chemical compound CC1(C)N=C(N)N=C(N)N1OCC1=CC=C(Cl)C(Cl)=C1 XDTNOYLBDDCJSK-UHFFFAOYSA-N 0.000 description 1
- ZOWYFYXTIWQBEP-UHFFFAOYSA-N 1-[(3,4-diethoxyphenyl)methyl]-6,7-diethoxyisoquinoline Chemical compound C1=C(OCC)C(OCC)=CC=C1CC1=NC=CC2=CC(OCC)=C(OCC)C=C12 ZOWYFYXTIWQBEP-UHFFFAOYSA-N 0.000 description 1
- PWZXTLHDGIBNLG-UHFFFAOYSA-N 1-[(3,4-dimethoxyphenyl)methyl]-6,7-dimethoxy-4-[[4-(2-methoxyphenyl)piperazin-1-yl]methyl]isoquinoline Chemical compound C1=C(OC)C(OC)=CC=C1CC(C1=CC(OC)=C(OC)C=C11)=NC=C1CN1CCN(C=2C(=CC=CC=2)OC)CC1 PWZXTLHDGIBNLG-UHFFFAOYSA-N 0.000 description 1
- BOVGTQGAOIONJV-BETUJISGSA-N 1-[(3ar,6as)-3,3a,4,5,6,6a-hexahydro-1h-cyclopenta[c]pyrrol-2-yl]-3-(4-methylphenyl)sulfonylurea Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1C[C@H]2CCC[C@H]2C1 BOVGTQGAOIONJV-BETUJISGSA-N 0.000 description 1
- ATEJLEVEJWUXMA-XGYZZIOMSA-N 1-[(3s,6s,8s,9s,10r,13s,14s,16r,17s)-3-hydroxy-6,10,13,16-tetramethyl-2,3,6,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C1([C@@H](C)C2)=C[C@@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(C)=O)[C@@]2(C)CC1 ATEJLEVEJWUXMA-XGYZZIOMSA-N 0.000 description 1
- GVALVWDQPXGPCE-UHFFFAOYSA-N 1-[(4-chlorophenoxy)methyl]-1,2,3,4-tetrahydroisoquinoline-6,7-diol Chemical compound C1=2C=C(O)C(O)=CC=2CCNC1COC1=CC=C(Cl)C=C1 GVALVWDQPXGPCE-UHFFFAOYSA-N 0.000 description 1
- YHDHSSBAHPSMPM-UHFFFAOYSA-N 1-[(4-chlorophenoxy)methyl]-3,4-dihydroisoquinoline Chemical compound C1=CC(Cl)=CC=C1OCC1=NCCC2=CC=CC=C12 YHDHSSBAHPSMPM-UHFFFAOYSA-N 0.000 description 1
- WFNAKBGANONZEQ-UHFFFAOYSA-N 1-[(4-chlorophenyl)-phenylmethyl]-4-methylpiperazine Chemical compound C1CN(C)CCN1C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 WFNAKBGANONZEQ-UHFFFAOYSA-N 0.000 description 1
- ABVFVJRTKMVJMV-XDHOZWIPSA-N 1-[(e)-2-[2-(4-chlorophenoxy)ethoxy]-2-(2,4-dichlorophenyl)ethenyl]imidazole Chemical compound C1=CC(Cl)=CC=C1OCCO\C(C=1C(=CC(Cl)=CC=1)Cl)=C\N1C=NC=C1 ABVFVJRTKMVJMV-XDHOZWIPSA-N 0.000 description 1
- OQCYTSHIQNPJIC-QURGRASLSA-N 1-[(e)-2-bromo-1,2-diphenylethenyl]-4-ethylbenzene Chemical compound C1=CC(CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Br)C1=CC=CC=C1 OQCYTSHIQNPJIC-QURGRASLSA-N 0.000 description 1
- HIQONTNPQNNMST-UBKPWBPPSA-N 1-[(e)-[5-(3,4-dichlorophenyl)furan-2-yl]methylideneamino]imidazolidine-2,4-dione Chemical compound C1=C(Cl)C(Cl)=CC=C1C(O1)=CC=C1\C=N\N1C(=O)NC(=O)C1 HIQONTNPQNNMST-UBKPWBPPSA-N 0.000 description 1
- SEGCNGONCZQFDW-OMCISZLKSA-N 1-[(e)-[5-(4-bromophenyl)-1,3-oxazol-2-yl]methylideneamino]imidazolidine-2,4-dione Chemical compound C1=CC(Br)=CC=C1C(O1)=CN=C1\C=N\N1C(=O)NC(=O)C1 SEGCNGONCZQFDW-OMCISZLKSA-N 0.000 description 1
- OZOMQRBLCMDCEG-CHHVJCJISA-N 1-[(z)-[5-(4-nitrophenyl)furan-2-yl]methylideneamino]imidazolidine-2,4-dione Chemical compound C1=CC([N+](=O)[O-])=CC=C1C(O1)=CC=C1\C=N/N1C(=O)NC(=O)C1 OZOMQRBLCMDCEG-CHHVJCJISA-N 0.000 description 1
- FITWYAUFKJXWPL-UHFFFAOYSA-N 1-[1-(2-methylpropoxy)-3-(1-prop-1-ynylcyclohexyl)oxypropan-2-yl]pyrrolidine Chemical compound C1CCCN1C(COCC(C)C)COC1(C#CC)CCCCC1 FITWYAUFKJXWPL-UHFFFAOYSA-N 0.000 description 1
- BFXRCMJRKTUNIQ-UHFFFAOYSA-N 1-[1-(2-methylpropoxy)-3-(2-phenylbut-3-yn-2-yloxy)propan-2-yl]pyrrolidine Chemical compound C1CCCN1C(COCC(C)C)COC(C)(C#C)C1=CC=CC=C1 BFXRCMJRKTUNIQ-UHFFFAOYSA-N 0.000 description 1
- JTUQXGZRVLWBCR-UHFFFAOYSA-N 1-[1-[2-(phenylmethyl)phenoxy]propan-2-yl]piperidine Chemical compound C1CCCCN1C(C)COC1=CC=CC=C1CC1=CC=CC=C1 JTUQXGZRVLWBCR-UHFFFAOYSA-N 0.000 description 1
- ZCJYUTQZBAIHBS-UHFFFAOYSA-N 1-[2-(2,4-dichlorophenyl)-2-{[4-(phenylsulfanyl)benzyl]oxy}ethyl]imidazole Chemical compound ClC1=CC(Cl)=CC=C1C(OCC=1C=CC(SC=2C=CC=CC=2)=CC=1)CN1C=NC=C1 ZCJYUTQZBAIHBS-UHFFFAOYSA-N 0.000 description 1
- RPZOFMHRRHHDPZ-UHFFFAOYSA-N 1-[2-(2-cyanoaziridin-1-yl)propan-2-yl]aziridine-2-carboxamide Chemical compound C1C(C(N)=O)N1C(C)(C)N1CC1C#N RPZOFMHRRHHDPZ-UHFFFAOYSA-N 0.000 description 1
- HLUGSJZJQJZYAE-UHFFFAOYSA-N 1-[2-(3,4-dimethoxyphenyl)ethylamino]-3-[6-(2-propan-2-ylidenehydrazinyl)pyridazin-3-yl]oxypropan-2-ol Chemical compound C1=C(OC)C(OC)=CC=C1CCNCC(O)COC1=CC=C(NN=C(C)C)N=N1 HLUGSJZJQJZYAE-UHFFFAOYSA-N 0.000 description 1
- MLHFXOOXBZMGSH-UHFFFAOYSA-N 1-[2-(4-bromophenyl)-2-phenylethyl]imidazole Chemical compound C1=CC(Br)=CC=C1C(C=1C=CC=CC=1)CN1C=NC=C1 MLHFXOOXBZMGSH-UHFFFAOYSA-N 0.000 description 1
- SZQLTQPHXXXMNC-UHFFFAOYSA-N 1-[2-(6,11-dihydrobenzo[c][1]benzoxepin-11-yl)ethyl-methylamino]-3-phenoxypropan-2-ol Chemical compound C12=CC=CC=C2COC2=CC=CC=C2C1CCN(C)CC(O)COC1=CC=CC=C1 SZQLTQPHXXXMNC-UHFFFAOYSA-N 0.000 description 1
- PZBPKYOVPCNPJY-UHFFFAOYSA-N 1-[2-(allyloxy)-2-(2,4-dichlorophenyl)ethyl]imidazole Chemical compound ClC1=CC(Cl)=CC=C1C(OCC=C)CN1C=NC=C1 PZBPKYOVPCNPJY-UHFFFAOYSA-N 0.000 description 1
- DHCZIHSQOICDAY-UHFFFAOYSA-N 1-[2-(diethylamino)ethoxy]-3-(2-methoxyphenoxy)propan-2-ol Chemical compound CCN(CC)CCOCC(O)COC1=CC=CC=C1OC DHCZIHSQOICDAY-UHFFFAOYSA-N 0.000 description 1
- KIPJSVIZACGFEX-UHFFFAOYSA-N 1-[2-[(2-chlorophenyl)-phenylmethyl]sulfanylethyl]-4-[(2-methylphenyl)methyl]piperazine Chemical compound CC1=CC=CC=C1CN1CCN(CCSC(C=2C=CC=CC=2)C=2C(=CC=CC=2)Cl)CC1 KIPJSVIZACGFEX-UHFFFAOYSA-N 0.000 description 1
- FLNXBVJLPJNOSI-UHFFFAOYSA-N 1-[2-[(4-chlorophenyl)-phenylmethoxy]ethyl]piperidine Chemical compound C1=CC(Cl)=CC=C1C(C=1C=CC=CC=1)OCCN1CCCCC1 FLNXBVJLPJNOSI-UHFFFAOYSA-N 0.000 description 1
- ATQREMXMJIWUIN-UHFFFAOYSA-N 1-[2-[ethyl-(2-hydroxy-2-methylpropyl)amino]ethylamino]-4-methylthioxanthen-9-one Chemical compound S1C2=CC=CC=C2C(=O)C2=C1C(C)=CC=C2NCCN(CC)CC(C)(C)O ATQREMXMJIWUIN-UHFFFAOYSA-N 0.000 description 1
- DIYPCWKHSODVAP-UHFFFAOYSA-N 1-[3-(2,5-dioxopyrrol-1-yl)benzoyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)C1=CC=CC(N2C(C=CC2=O)=O)=C1 DIYPCWKHSODVAP-UHFFFAOYSA-N 0.000 description 1
- KZZIVOLMJPSDEP-UHFFFAOYSA-N 1-[3-(4-fluorophenyl)-3-pyridin-2-ylpropyl]-2-[3-(1h-imidazol-5-yl)propyl]guanidine Chemical compound C=1N=CNC=1CCCN=C(N)NCCC(C=1N=CC=CC=1)C1=CC=C(F)C=C1 KZZIVOLMJPSDEP-UHFFFAOYSA-N 0.000 description 1
- DPQPWAGLQYSJRP-UHFFFAOYSA-M 1-[3-(cyclohexen-1-yl)-3-phenylpropyl]-1-methylpiperidin-1-ium;methyl sulfate Chemical compound COS([O-])(=O)=O.C=1CCCCC=1C(C=1C=CC=CC=1)CC[N+]1(C)CCCCC1 DPQPWAGLQYSJRP-UHFFFAOYSA-M 0.000 description 1
- XKSOTQXTPALQMY-UHFFFAOYSA-N 1-[3-[(4-azidophenyl)disulfanyl]propanoyloxy]-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)CCSSC1=CC=C(N=[N+]=[N-])C=C1 XKSOTQXTPALQMY-UHFFFAOYSA-N 0.000 description 1
- XKQYCEFPFNDDSJ-UHFFFAOYSA-N 1-[3-[2-[(4-azido-2-hydroxybenzoyl)amino]ethyldisulfanyl]propanoyloxy]-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound OC1=CC(N=[N+]=[N-])=CC=C1C(=O)NCCSSCCC(=O)ON1C(=O)C(S(O)(=O)=O)CC1=O XKQYCEFPFNDDSJ-UHFFFAOYSA-N 0.000 description 1
- VOTJUWBJENROFB-UHFFFAOYSA-N 1-[3-[[3-(2,5-dioxo-3-sulfopyrrolidin-1-yl)oxy-3-oxopropyl]disulfanyl]propanoyloxy]-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)CCSSCCC(=O)ON1C(=O)C(S(O)(=O)=O)CC1=O VOTJUWBJENROFB-UHFFFAOYSA-N 0.000 description 1
- BWMMRMDCODVQBX-UHFFFAOYSA-N 1-[4,7-dimethoxy-6-(2-pyrrolidin-1-ylethoxy)-1-benzofuran-5-yl]-3-methylurea Chemical compound COC1=C2OC=CC2=C(OC)C(NC(=O)NC)=C1OCCN1CCCC1 BWMMRMDCODVQBX-UHFFFAOYSA-N 0.000 description 1
- WQQBUTMELIQJNY-UHFFFAOYSA-N 1-[4-(2,5-dioxo-3-sulfopyrrolidin-1-yl)oxy-2,3-dihydroxy-4-oxobutanoyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1CC(S(O)(=O)=O)C(=O)N1OC(=O)C(O)C(O)C(=O)ON1C(=O)CC(S(O)(=O)=O)C1=O WQQBUTMELIQJNY-UHFFFAOYSA-N 0.000 description 1
- KSQCNASWXSCJTD-UHFFFAOYSA-N 1-[4-(2-methoxyphenyl)piperazin-1-yl]-3-(3,4,5-trimethoxyphenoxy)propan-2-ol Chemical compound COC1=CC=CC=C1N1CCN(CC(O)COC=2C=C(OC)C(OC)=C(OC)C=2)CC1 KSQCNASWXSCJTD-UHFFFAOYSA-N 0.000 description 1
- CULQNACJHGHAER-UHFFFAOYSA-N 1-[4-[(2-iodoacetyl)amino]benzoyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)C1=CC=C(NC(=O)CI)C=C1 CULQNACJHGHAER-UHFFFAOYSA-N 0.000 description 1
- KMICWMXRUOETFR-UHFFFAOYSA-N 1-[4-[2-(diethylamino)ethoxy]phenyl]-3-phenylpropan-1-one Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(=O)CCC1=CC=CC=C1 KMICWMXRUOETFR-UHFFFAOYSA-N 0.000 description 1
- XQFRJNBWHJMXHO-UHFFFAOYSA-N 1-[4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-iodopyrimidine-2,4-dione Chemical compound C1C(O)C(CO)OC1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-UHFFFAOYSA-N 0.000 description 1
- UPNUQQDXHCUWSG-UHFFFAOYSA-N 1-[6-(4-azido-2-nitroanilino)hexanoyloxy]-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)CCCCCNC1=CC=C(N=[N+]=[N-])C=C1[N+]([O-])=O UPNUQQDXHCUWSG-UHFFFAOYSA-N 0.000 description 1
- INJPEINGXBVKEB-RTBURBONSA-N 1-[[(2r,3r)-3-[(2,6-difluorophenyl)methoxy]-5-fluoro-2,3-dihydro-1-benzothiophen-2-yl]methyl]imidazole Chemical compound C([C@H]1SC2=CC=C(C=C2[C@H]1OCC=1C(=CC=CC=1F)F)F)N1C=CN=C1 INJPEINGXBVKEB-RTBURBONSA-N 0.000 description 1
- OCAPBUJLXMYKEJ-UHFFFAOYSA-N 1-[biphenyl-4-yl(phenyl)methyl]imidazole Chemical compound C1=NC=CN1C(C=1C=CC(=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 OCAPBUJLXMYKEJ-UHFFFAOYSA-N 0.000 description 1
- GZIZSEZACLNKGZ-UHFFFAOYSA-N 1-amino-3-decoxypropan-2-ol Chemical compound CCCCCCCCCCOCC(O)CN GZIZSEZACLNKGZ-UHFFFAOYSA-N 0.000 description 1
- NILQLFBWTXNUOE-UHFFFAOYSA-N 1-aminocyclopentanecarboxylic acid Chemical compound OC(=O)C1(N)CCCC1 NILQLFBWTXNUOE-UHFFFAOYSA-N 0.000 description 1
- AHKAOMZZTQULDS-UHFFFAOYSA-N 1-azabicyclo[2.2.2]octan-3-yl benzoate Chemical compound C1N(CC2)CCC2C1OC(=O)C1=CC=CC=C1 AHKAOMZZTQULDS-UHFFFAOYSA-N 0.000 description 1
- GNQQJMWZCJAUJT-RSGUCCNWSA-N 1-benzhydryl-4-[(e)-3-phenylprop-2-enyl]piperazine;2-(4-chlorophenoxy)-2-methylpropanoic acid Chemical compound OC(=O)C(C)(C)OC1=CC=C(Cl)C=C1.C1CN(C(C=2C=CC=CC=2)C=2C=CC=CC=2)CCN1C\C=C\C1=CC=CC=C1 GNQQJMWZCJAUJT-RSGUCCNWSA-N 0.000 description 1
- BRCYAOFSVFIBPV-UHFFFAOYSA-N 1-benzo[b][1]benzothiepin-5-yl-n,n-dimethylmethanamine Chemical compound CN(C)CC1=CC2=CC=CC=C2SC2=CC=CC=C12 BRCYAOFSVFIBPV-UHFFFAOYSA-N 0.000 description 1
- LHVNPTMRAQQPID-UHFFFAOYSA-N 1-benzyl-2,3,4,9-tetrahydro-1h-pyrido[3,4-b]indole Chemical compound N1CCC(C2=CC=CC=C2N2)=C2C1CC1=CC=CC=C1 LHVNPTMRAQQPID-UHFFFAOYSA-N 0.000 description 1
- VRJCKJKZCZLXBK-UHFFFAOYSA-N 1-benzyl-3-(2-pyridin-4-ylethyl)indole Chemical compound C=1N(CC=2C=CC=CC=2)C2=CC=CC=C2C=1CCC1=CC=NC=C1 VRJCKJKZCZLXBK-UHFFFAOYSA-N 0.000 description 1
- PUDJZXFLQHLFGS-UHFFFAOYSA-N 1-benzylcyclohepta[d]imidazol-2-one Chemical compound O=C1N=C2C=CC=CC=C2N1CC1=CC=CC=C1 PUDJZXFLQHLFGS-UHFFFAOYSA-N 0.000 description 1
- CPZHDDGPHQRMEJ-UHFFFAOYSA-N 1-butyl-3-[1-(6,7-dimethoxyquinazolin-4-yl)piperidin-4-yl]urea Chemical compound C1CC(NC(=O)NCCCC)CCN1C1=NC=NC2=CC(OC)=C(OC)C=C12 CPZHDDGPHQRMEJ-UHFFFAOYSA-N 0.000 description 1
- VEPZOLKTNZOTTQ-UHFFFAOYSA-N 1-butyl-n-(2,4,6-trimethylphenyl)pyrrolidine-2-carboxamide Chemical compound CCCCN1CCCC1C(=O)NC1=C(C)C=C(C)C=C1C VEPZOLKTNZOTTQ-UHFFFAOYSA-N 0.000 description 1
- MKSYSNRVVYYBIZ-UHFFFAOYSA-N 1-butyl-n-(3,4-dichlorophenyl)pyrrolidin-2-imine Chemical compound CCCCN1CCCC1=NC1=CC=C(Cl)C(Cl)=C1 MKSYSNRVVYYBIZ-UHFFFAOYSA-N 0.000 description 1
- KEDVUOWPLAHMLZ-UHFFFAOYSA-N 1-cyano-3-[2-[(5-methyl-1h-imidazol-4-yl)methylsulfanyl]ethyl]-2-prop-2-ynylguanidine Chemical compound CC=1NC=NC=1CSCCNC(NC#N)=NCC#C KEDVUOWPLAHMLZ-UHFFFAOYSA-N 0.000 description 1
- DZNUOUOIPRQTTB-UHFFFAOYSA-N 1-cyclohexyl-2-methylpropan-1-ol Chemical compound CC(C)C(O)C1CCCCC1 DZNUOUOIPRQTTB-UHFFFAOYSA-N 0.000 description 1
- LLJFMFZYVVLQKT-UHFFFAOYSA-N 1-cyclohexyl-3-[4-[2-(7-methoxy-4,4-dimethyl-1,3-dioxo-2-isoquinolinyl)ethyl]phenyl]sulfonylurea Chemical compound C=1C(OC)=CC=C(C(C2=O)(C)C)C=1C(=O)N2CCC(C=C1)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 LLJFMFZYVVLQKT-UHFFFAOYSA-N 0.000 description 1
- XTOBSLUVJNOXGB-UHFFFAOYSA-N 1-cyclooctyl-3-(4-methylphenyl)sulfonylurea Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NC1CCCCCCC1 XTOBSLUVJNOXGB-UHFFFAOYSA-N 0.000 description 1
- NQTSTBMCCAVWOS-UHFFFAOYSA-N 1-dimethoxyphosphoryl-3-phenoxypropan-2-one Chemical compound COP(=O)(OC)CC(=O)COC1=CC=CC=C1 NQTSTBMCCAVWOS-UHFFFAOYSA-N 0.000 description 1
- YETULFFXNIHQLK-UHFFFAOYSA-N 1-ethynyl-4-(2-fluorophenyl)benzene Chemical compound FC1=CC=CC=C1C1=CC=C(C#C)C=C1 YETULFFXNIHQLK-UHFFFAOYSA-N 0.000 description 1
- UIXYQZIHFQKFOZ-UHFFFAOYSA-N 1-methyl-3-[2-[4-[(4-methylcyclohexyl)carbamoylsulfamoyl]phenyl]ethyl]-1-pyridin-2-ylurea Chemical compound C1CC(C)CCC1NC(=O)NS(=O)(=O)C(C=C1)=CC=C1CCNC(=O)N(C)C1=CC=CC=N1 UIXYQZIHFQKFOZ-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- CRKZWPJRHFAGCJ-UHFFFAOYSA-N 1-phenoxy-3-[2-[(1,3,5-trimethylpyrazol-4-yl)amino]ethylamino]propan-2-ol Chemical compound CC1=NN(C)C(C)=C1NCCNCC(O)COC1=CC=CC=C1 CRKZWPJRHFAGCJ-UHFFFAOYSA-N 0.000 description 1
- CFJMRBQWBDQYMK-UHFFFAOYSA-N 1-phenyl-1-cyclopentanecarboxylic acid 2-[2-(diethylamino)ethoxy]ethyl ester Chemical compound C=1C=CC=CC=1C1(C(=O)OCCOCCN(CC)CC)CCCC1 CFJMRBQWBDQYMK-UHFFFAOYSA-N 0.000 description 1
- OHHIAWWXDMYFGP-UHFFFAOYSA-N 1-phenyl-2-(pyrimidin-2-ylamino)ethanol Chemical compound C=1C=CC=CC=1C(O)CNC1=NC=CC=N1 OHHIAWWXDMYFGP-UHFFFAOYSA-N 0.000 description 1
- VIAYOUBFHWUVLW-FMIVXFBMSA-N 1-phenyl-3-[2-[[4-[(e)-3-phenylprop-2-enyl]piperazin-1-yl]methyl]benzimidazol-1-yl]propan-1-one Chemical compound C=1C=CC=CC=1C(=O)CCN(C1=CC=CC=C1N=1)C=1CN(CC1)CCN1C\C=C\C1=CC=CC=C1 VIAYOUBFHWUVLW-FMIVXFBMSA-N 0.000 description 1
- TYGVJQQISVEFSH-UHFFFAOYSA-N 1-phenyl-3-piperidin-1-ylpyrrolidin-2-one Chemical compound O=C1C(N2CCCCC2)CCN1C1=CC=CC=C1 TYGVJQQISVEFSH-UHFFFAOYSA-N 0.000 description 1
- LEZWWPYKPKIXLL-UHFFFAOYSA-N 1-{2-(4-chlorobenzyloxy)-2-(2,4-dichlorophenyl)ethyl}imidazole Chemical compound C1=CC(Cl)=CC=C1COC(C=1C(=CC(Cl)=CC=1)Cl)CN1C=NC=C1 LEZWWPYKPKIXLL-UHFFFAOYSA-N 0.000 description 1
- HLBRHTFSMMEHPK-UHFFFAOYSA-N 10-(1-methylpiperidin-4-ylidene)-9h-anthracen-9-ol Chemical compound C1CN(C)CCC1=C1C2=CC=CC=C2C(O)C2=CC=CC=C21 HLBRHTFSMMEHPK-UHFFFAOYSA-N 0.000 description 1
- VRKHTAYPELFGPP-UHFFFAOYSA-N 10-[(1,3-dimethylpyrrolidin-3-yl)methyl]phenothiazine Chemical compound C1N(C)CCC1(C)CN1C2=CC=CC=C2SC2=CC=CC=C21 VRKHTAYPELFGPP-UHFFFAOYSA-N 0.000 description 1
- BPEQXNIRIPLFRW-UHFFFAOYSA-N 10-[1-[2-[2-(2-hydroxyethoxy)ethoxy]ethyl]piperidin-4-ylidene]-5h-benzo[1,2]cyclohepta[3,4-b]thiophen-4-one Chemical compound C1CN(CCOCCOCCO)CCC1=C1C2=CC=CC=C2CC(=O)C2=C1C=CS2 BPEQXNIRIPLFRW-UHFFFAOYSA-N 0.000 description 1
- KRJLZXUYKFHTSN-UHFFFAOYSA-L 10-[dimethyl-[2-(5-methyl-2-propan-2-ylphenoxy)ethyl]azaniumyl]decyl-dimethyl-[2-(5-methyl-2-propan-2-ylphenoxy)ethyl]azanium;dibromide Chemical compound [Br-].[Br-].CC(C)C1=CC=C(C)C=C1OCC[N+](C)(C)CCCCCCCCCC[N+](C)(C)CCOC1=CC(C)=CC=C1C(C)C KRJLZXUYKFHTSN-UHFFFAOYSA-L 0.000 description 1
- AHZXFRRDQXXPJD-UHFFFAOYSA-N 10-butanoyl-1,8-dihydroxy-10h-anthracen-9-one Chemical compound C1=CC=C2C(C(=O)CCC)C3=CC=CC(O)=C3C(=O)C2=C1O AHZXFRRDQXXPJD-UHFFFAOYSA-N 0.000 description 1
- FWBSWSRNGHRDFZ-UHFFFAOYSA-N 10-methoxy-2-[2-[4-[1-[2-(10-methoxy-7h-pyrido[4,3-c]carbazol-2-ium-2-yl)ethyl]piperidin-4-yl]piperidin-1-yl]ethyl]-7h-pyrido[4,3-c]carbazol-2-ium;dichloride Chemical compound [Cl-].[Cl-].N1C2=CC=C(OC)C=C2C(C2=C3)=C1C=CC2=CC=[N+]3CCN(CC1)CCC1C(CC1)CCN1CC[N+]1=CC2=C(C=3C(=CC=C(C=3)OC)N3)C3=CC=C2C=C1 FWBSWSRNGHRDFZ-UHFFFAOYSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- VVOIQBFMTVCINR-WWMZEODYSA-N 11-deoxycorticosterone pivalate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)COC(=O)C(C)(C)C)[C@@]1(C)CC2 VVOIQBFMTVCINR-WWMZEODYSA-N 0.000 description 1
- WHBHBVVOGNECLV-OBQKJFGGSA-N 11-deoxycortisol Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 WHBHBVVOGNECLV-OBQKJFGGSA-N 0.000 description 1
- HMZDSBSMNLBGIW-UHFFFAOYSA-N 11-oxopyrido[2,1-b]quinazoline-2-carboxylic acid Chemical compound C1=CC=CN2C(=O)C3=CC(C(=O)O)=CC=C3N=C21 HMZDSBSMNLBGIW-UHFFFAOYSA-N 0.000 description 1
- HXQAPLNYYFQSFU-UHFFFAOYSA-N 11h-dibenzo[1,2-a:1',2'-e][7]annulene-11-carboxamide Chemical compound C1=CC2=CC=CC=C2C(C(=O)N)C2=CC=CC=C21 HXQAPLNYYFQSFU-UHFFFAOYSA-N 0.000 description 1
- OOSYXMPLIGBCDA-UHFFFAOYSA-N 13-cyclopentyl-n-[4-[4-(13-cyclopentyltridecanoylamino)phenyl]sulfonylphenyl]tridecanamide Chemical compound C=1C=C(S(=O)(=O)C=2C=CC(NC(=O)CCCCCCCCCCCCC3CCCC3)=CC=2)C=CC=1NC(=O)CCCCCCCCCCCCC1CCCC1 OOSYXMPLIGBCDA-UHFFFAOYSA-N 0.000 description 1
- XSGQFHNPNWBVPT-VFXMVCAWSA-N 15-methyl-15R-PGE2 Chemical compound CCCCC[C@@](C)(O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XSGQFHNPNWBVPT-VFXMVCAWSA-N 0.000 description 1
- PROQIPRRNZUXQM-ZMSHIADSSA-N 16beta-hydroxyestradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H]([C@@H](O)C4)O)[C@@H]4[C@@H]3CCC2=C1 PROQIPRRNZUXQM-ZMSHIADSSA-N 0.000 description 1
- BFPYWIDHMRZLRN-UHFFFAOYSA-N 17alpha-ethynyl estradiol Natural products OC1=CC=C2C3CCC(C)(C(CC4)(O)C#C)C4C3CCC2=C1 BFPYWIDHMRZLRN-UHFFFAOYSA-N 0.000 description 1
- MPDGHEJMBKOTSU-YKLVYJNSSA-N 18beta-glycyrrhetic acid Chemical compound C([C@H]1C2=CC(=O)[C@H]34)[C@@](C)(C(O)=O)CC[C@]1(C)CC[C@@]2(C)[C@]4(C)CC[C@@H]1[C@]3(C)CC[C@H](O)C1(C)C MPDGHEJMBKOTSU-YKLVYJNSSA-N 0.000 description 1
- NMPZCCZXCOMSDQ-XVFCMESISA-N 2',3'-cyclic CMP Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H]2OP(O)(=O)O[C@@H]2[C@@H](CO)O1 NMPZCCZXCOMSDQ-XVFCMESISA-N 0.000 description 1
- MEZZCSHVIGVWFI-UHFFFAOYSA-N 2,2'-Dihydroxy-4-methoxybenzophenone Chemical compound OC1=CC(OC)=CC=C1C(=O)C1=CC=CC=C1O MEZZCSHVIGVWFI-UHFFFAOYSA-N 0.000 description 1
- DJKWDKJYFYUEBQ-UHFFFAOYSA-N 2,2,9,9-tetramethyldecane-1,10-diol Chemical compound OCC(C)(C)CCCCCCC(C)(C)CO DJKWDKJYFYUEBQ-UHFFFAOYSA-N 0.000 description 1
- MLVKMSCFQIQULM-UHFFFAOYSA-N 2,2-dichloro-n-[3-chloro-1-(4-chlorophenyl)-1-oxopropan-2-yl]acetamide Chemical compound ClC(Cl)C(=O)NC(CCl)C(=O)C1=CC=C(Cl)C=C1 MLVKMSCFQIQULM-UHFFFAOYSA-N 0.000 description 1
- PIPWSBOFSUJCCO-UHFFFAOYSA-N 2,2-dimethylpiperazine Chemical compound CC1(C)CNCCN1 PIPWSBOFSUJCCO-UHFFFAOYSA-N 0.000 description 1
- SCVCXWHEHAKJCG-UHFFFAOYSA-N 2,3,4,6,7,11b-hexahydro-1h-pyrazino[2,1-a]isoquinoline Chemical compound C1CC2=CC=CC=C2C2N1CCNC2 SCVCXWHEHAKJCG-UHFFFAOYSA-N 0.000 description 1
- NYIZXMGNIUSNKL-UHFFFAOYSA-N 2,3-diacetyloxybenzoic acid Chemical group CC(=O)OC1=CC=CC(C(O)=O)=C1OC(C)=O NYIZXMGNIUSNKL-UHFFFAOYSA-N 0.000 description 1
- SWYJYGCPTGKBDS-UHFFFAOYSA-N 2,3-dihydroxypropyl 2-(3-chloro-2-methylanilino)pyridine-3-carboxylate Chemical compound CC1=C(Cl)C=CC=C1NC1=NC=CC=C1C(=O)OCC(O)CO SWYJYGCPTGKBDS-UHFFFAOYSA-N 0.000 description 1
- STDYWHYUOSSCBO-UHFFFAOYSA-N 2,3-dimethyl-4-phenyl-4,5-dihydro-1,3-benzodiazepine Chemical compound C1C2=CC=CC=C2N=C(C)N(C)C1C1=CC=CC=C1 STDYWHYUOSSCBO-UHFFFAOYSA-N 0.000 description 1
- IYOLBFFHPZOQGW-UHFFFAOYSA-N 2,4-dichloro-3,5-dimethylphenol Chemical compound CC1=CC(O)=C(Cl)C(C)=C1Cl IYOLBFFHPZOQGW-UHFFFAOYSA-N 0.000 description 1
- RPAJWWXZIQJVJF-UHFFFAOYSA-N 2,4-dichloro-6-(3,5-dichloro-2-hydroxyphenyl)sulfinylphenol Chemical compound OC1=C(Cl)C=C(Cl)C=C1S(=O)C1=CC(Cl)=CC(Cl)=C1O RPAJWWXZIQJVJF-UHFFFAOYSA-N 0.000 description 1
- BOMZMNZEXMAQQW-UHFFFAOYSA-N 2,5,11-trimethyl-6h-pyrido[4,3-b]carbazol-2-ium-9-ol;acetate Chemical compound CC([O-])=O.C[N+]1=CC=C2C(C)=C(NC=3C4=CC(O)=CC=3)C4=C(C)C2=C1 BOMZMNZEXMAQQW-UHFFFAOYSA-N 0.000 description 1
- FXMWUTGUCAKGQL-UHFFFAOYSA-N 2,5-dimethoxy-4-bromoamphetamine Chemical compound COC1=CC(CC(C)N)=C(OC)C=C1Br FXMWUTGUCAKGQL-UHFFFAOYSA-N 0.000 description 1
- ASNTZYQMIUCEBV-UHFFFAOYSA-N 2,5-dioxo-1-[6-[3-(pyridin-2-yldisulfanyl)propanoylamino]hexanoyloxy]pyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)CCCCCNC(=O)CCSSC1=CC=CC=N1 ASNTZYQMIUCEBV-UHFFFAOYSA-N 0.000 description 1
- CYJQCYXRNNCURD-UHFFFAOYSA-N 2,8-dimethyl-1,3,4,4a,5,9b-hexahydropyrido[4,3-b]indole Chemical compound N1C2=CC=C(C)C=C2C2C1CCN(C)C2 CYJQCYXRNNCURD-UHFFFAOYSA-N 0.000 description 1
- OAOFTCGGCRBLAW-UHFFFAOYSA-M 2-(1,2,5-trimethylpyrrolidin-1-ium-1-yl)ethyl 2-hydroxy-2,2-diphenylacetate;bromide Chemical compound [Br-].CC1CCC(C)[N+]1(C)CCOC(=O)C(O)(C=1C=CC=CC=1)C1=CC=CC=C1 OAOFTCGGCRBLAW-UHFFFAOYSA-M 0.000 description 1
- AZCOEIZFVQEQCU-UHFFFAOYSA-N 2-(1-benzoyl-2-methylindol-3-yl)acetic acid Chemical compound CC1=C(CC(O)=O)C2=CC=CC=C2N1C(=O)C1=CC=CC=C1 AZCOEIZFVQEQCU-UHFFFAOYSA-N 0.000 description 1
- RPSOLZRELOLSFM-UHFFFAOYSA-N 2-(1-benzyl-5-methoxy-2-methylindol-3-yl)ethanamine Chemical compound CC1=C(CCN)C2=CC(OC)=CC=C2N1CC1=CC=CC=C1 RPSOLZRELOLSFM-UHFFFAOYSA-N 0.000 description 1
- PCLIRWBVOVZTOK-UHFFFAOYSA-M 2-(1-methylpyrrolidin-1-ium-1-yl)ethyl 2-hydroxy-2,2-diphenylacetate;iodide Chemical compound [I-].C=1C=CC=CC=1C(O)(C=1C=CC=CC=1)C(=O)OCC[N+]1(C)CCCC1 PCLIRWBVOVZTOK-UHFFFAOYSA-M 0.000 description 1
- JONFYSWOLCTYFK-UHFFFAOYSA-N 2-(2,3-dichloroanilino)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1NC1=CC=CC(Cl)=C1Cl JONFYSWOLCTYFK-UHFFFAOYSA-N 0.000 description 1
- IFMIBXMJNXNGHQ-UHFFFAOYSA-N 2-(2,3-dichloroanilino)pyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=CN=C1NC1=CC=CC(Cl)=C1Cl IFMIBXMJNXNGHQ-UHFFFAOYSA-N 0.000 description 1
- BCPFWSWROVXGQA-UHFFFAOYSA-N 2-(2,4,4-trimethylpentan-2-yl)guanidine Chemical compound CC(C)(C)CC(C)(C)N=C(N)N BCPFWSWROVXGQA-UHFFFAOYSA-N 0.000 description 1
- BPXIOWINQPQVOY-UHFFFAOYSA-N 2-(2-benzylphenoxy)-n,n-diethylethanamine Chemical compound CCN(CC)CCOC1=CC=CC=C1CC1=CC=CC=C1 BPXIOWINQPQVOY-UHFFFAOYSA-N 0.000 description 1
- XBNRCMFFZFCWMH-UHFFFAOYSA-N 2-(2-ethoxyethoxy)benzamide Chemical compound CCOCCOC1=CC=CC=C1C(N)=O XBNRCMFFZFCWMH-UHFFFAOYSA-N 0.000 description 1
- MYQXHLQMZLTSDB-UHFFFAOYSA-N 2-(2-ethyl-2,3-dihydro-1-benzofuran-5-yl)acetic acid Chemical compound OC(=O)CC1=CC=C2OC(CC)CC2=C1 MYQXHLQMZLTSDB-UHFFFAOYSA-N 0.000 description 1
- RATZLMXRALDSJW-UHFFFAOYSA-N 2-(2-ethyl-3H-benzofuran-2-yl)-4,5-dihydro-1H-imidazole Chemical compound C1C2=CC=CC=C2OC1(CC)C1=NCCN1 RATZLMXRALDSJW-UHFFFAOYSA-N 0.000 description 1
- RHDBQQXIWBILRU-UHFFFAOYSA-N 2-(2-methoxyphenyl)-3,3-diphenylprop-2-enoic acid Chemical compound COC1=CC=CC=C1C(C(O)=O)=C(C=1C=CC=CC=1)C1=CC=CC=C1 RHDBQQXIWBILRU-UHFFFAOYSA-N 0.000 description 1
- JOVXEDBYAWFQQX-UHFFFAOYSA-N 2-(2-methyl-5-nitroimidazol-1-yl)ethyl carbamate Chemical compound CC1=NC=C([N+]([O-])=O)N1CCOC(N)=O JOVXEDBYAWFQQX-UHFFFAOYSA-N 0.000 description 1
- FTBWWORDOHNJDB-UHFFFAOYSA-N 2-(3,4-dichlorophenyl)-3-methyl-1,1-dioxo-1,3-thiazinan-4-one Chemical compound O=S1(=O)CCC(=O)N(C)C1C1=CC=C(Cl)C(Cl)=C1 FTBWWORDOHNJDB-UHFFFAOYSA-N 0.000 description 1
- GXLVKSWWJRJBMC-VVSTWUKXSA-N 2-(3,4-dihydroxyphenyl)-5-hydroxy-7-(2-morpholin-4-ylethoxy)-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-[[(2r,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@H](OC=2C(C3=C(O)C=C(OCCN4CCOCC4)C=C3OC=2C=2C=C(O)C(O)=CC=2)=O)O1 GXLVKSWWJRJBMC-VVSTWUKXSA-N 0.000 description 1
- WWCVTBASZDLZAC-UHFFFAOYSA-N 2-(3,5-dimethoxyphenoxy)ethanol Chemical compound COC1=CC(OC)=CC(OCCO)=C1 WWCVTBASZDLZAC-UHFFFAOYSA-N 0.000 description 1
- LEBFQNREPOQDII-UHFFFAOYSA-N 2-(3-fluorobenzo[b][1]benzoxepin-5-yl)sulfanyl-n-methylethanamine Chemical compound CNCCSC1=CC2=CC=CC=C2OC2=CC=C(F)C=C12 LEBFQNREPOQDII-UHFFFAOYSA-N 0.000 description 1
- NJZMHNHROHOKDK-UHFFFAOYSA-N 2-(3-oxo-1-propan-2-ylpiperazin-1-ium-2-yl)acetate Chemical compound CC(C)[NH+]1CCNC(=O)C1CC([O-])=O NJZMHNHROHOKDK-UHFFFAOYSA-N 0.000 description 1
- UEIBTMGAIXZRBV-UHFFFAOYSA-N 2-(3-phenoxypropyl)guanidine Chemical compound NC(N)=NCCCOC1=CC=CC=C1 UEIBTMGAIXZRBV-UHFFFAOYSA-N 0.000 description 1
- ODZUWQAFWMLWCF-UHFFFAOYSA-N 2-(3-phenyl-1-benzofuran-7-yl)propanoic acid Chemical compound C=1OC=2C(C(C(O)=O)C)=CC=CC=2C=1C1=CC=CC=C1 ODZUWQAFWMLWCF-UHFFFAOYSA-N 0.000 description 1
- TXLHNFOLHRXMAU-UHFFFAOYSA-N 2-(4-benzylphenoxy)-n,n-diethylethanamine;hydron;chloride Chemical compound Cl.C1=CC(OCCN(CC)CC)=CC=C1CC1=CC=CC=C1 TXLHNFOLHRXMAU-UHFFFAOYSA-N 0.000 description 1
- LHQKYSCQIUUKDH-UHFFFAOYSA-N 2-(4-butoxyphenoxy)-n-(2-methoxyphenyl)-n-(2-pyrrolidin-1-ylethyl)acetamide Chemical compound C1=CC(OCCCC)=CC=C1OCC(=O)N(C=1C(=CC=CC=1)OC)CCN1CCCC1 LHQKYSCQIUUKDH-UHFFFAOYSA-N 0.000 description 1
- ADIBYIWFBDMKNY-UHFFFAOYSA-N 2-(4-butylanilino)pyridine-3-carboxylic acid Chemical compound C1=CC(CCCC)=CC=C1NC1=NC=CC=C1C(O)=O ADIBYIWFBDMKNY-UHFFFAOYSA-N 0.000 description 1
- 239000003315 2-(4-chlorophenoxy)-2-methylpropanoic acid Substances 0.000 description 1
- ZSZFUDFOPOMEET-UHFFFAOYSA-N 2-(4-chlorophenyl)-2-[2,6-dichloro-4-(3,5-dioxo-1,2,4-triazin-2-yl)phenyl]acetonitrile Chemical compound C1=CC(Cl)=CC=C1C(C#N)C1=C(Cl)C=C(N2C(NC(=O)C=N2)=O)C=C1Cl ZSZFUDFOPOMEET-UHFFFAOYSA-N 0.000 description 1
- DTCYNRIBBFRFNN-UHFFFAOYSA-N 2-(4-oxaldehydoylphenyl)-2-oxoacetaldehyde Chemical compound O=CC(=O)C1=CC=C(C(=O)C=O)C=C1 DTCYNRIBBFRFNN-UHFFFAOYSA-N 0.000 description 1
- TUMWSYRTKGBCAG-UHFFFAOYSA-N 2-(5-benzyl-6-sulfanylidene-1,3,5-thiadiazinan-3-yl)acetic acid Chemical compound C1N(CC(=O)O)CSC(=S)N1CC1=CC=CC=C1 TUMWSYRTKGBCAG-UHFFFAOYSA-N 0.000 description 1
- BYUAWKXUYPHXSR-UHFFFAOYSA-N 2-(8-chloro-6,11-dihydro-5h-benzo[1,2]cyclohepta[2,4-b]pyridin-11-yl)-n,n-dimethylethanamine Chemical compound C1CC2=CC=CN=C2C(CCN(C)C)C2=CC=C(Cl)C=C21 BYUAWKXUYPHXSR-UHFFFAOYSA-N 0.000 description 1
- KWGCXQZMMAVJTB-UHFFFAOYSA-N 2-(8-chlorodibenzofuran-3-yl)propanoic acid Chemical compound C1=C(Cl)C=C2C3=CC=C(C(C(O)=O)C)C=C3OC2=C1 KWGCXQZMMAVJTB-UHFFFAOYSA-N 0.000 description 1
- LRXFKKPEBXIPMW-UHFFFAOYSA-N 2-(9h-fluoren-2-yl)propanoic acid Chemical compound C1=CC=C2C3=CC=C(C(C(O)=O)C)C=C3CC2=C1 LRXFKKPEBXIPMW-UHFFFAOYSA-N 0.000 description 1
- DMEHEIWCWDNUBJ-UHFFFAOYSA-N 2-(cyclopropylmethyl)-5,6-bis(4-methoxyphenyl)-1,2,4-triazin-3-one Chemical compound C1=CC(OC)=CC=C1C(C(=NC1=O)C=2C=CC(OC)=CC=2)=NN1CC1CC1 DMEHEIWCWDNUBJ-UHFFFAOYSA-N 0.000 description 1
- UTTZVFFEPWFVRY-JZJYNLBNSA-N 2-(diethylamino)ethyl (z)-2,3-diphenylprop-2-enoate Chemical compound C=1C=CC=CC=1/C(C(=O)OCCN(CC)CC)=C/C1=CC=CC=C1 UTTZVFFEPWFVRY-JZJYNLBNSA-N 0.000 description 1
- DIDYGLSKVUKRRP-UHFFFAOYSA-N 2-(diethylamino)ethyl 2,2-diphenylpropanoate Chemical compound C=1C=CC=CC=1C(C)(C(=O)OCCN(CC)CC)C1=CC=CC=C1 DIDYGLSKVUKRRP-UHFFFAOYSA-N 0.000 description 1
- QXNKAUNWERFBBV-UHFFFAOYSA-N 2-(diethylamino)ethyl 2-hydroxybenzoate Chemical compound CCN(CC)CCOC(=O)C1=CC=CC=C1O QXNKAUNWERFBBV-UHFFFAOYSA-N 0.000 description 1
- BSRXEULOHCMVFG-UHFFFAOYSA-N 2-(diethylamino)ethyl 4-benzylpiperidine-1-carboxylate Chemical compound C1CN(C(=O)OCCN(CC)CC)CCC1CC1=CC=CC=C1 BSRXEULOHCMVFG-UHFFFAOYSA-N 0.000 description 1
- YDUHWSSRUBOPKZ-UHFFFAOYSA-N 2-(diethylamino)ethyl n,n-diphenylcarbamate Chemical compound C=1C=CC=CC=1N(C(=O)OCCN(CC)CC)C1=CC=CC=C1 YDUHWSSRUBOPKZ-UHFFFAOYSA-N 0.000 description 1
- DENMGZODXQRYAR-UHFFFAOYSA-N 2-(dimethylamino)ethanethiol Chemical compound CN(C)CCS DENMGZODXQRYAR-UHFFFAOYSA-N 0.000 description 1
- TXUGZLRUFAAHAO-LFIBNONCSA-N 2-(dimethylamino)ethyl 2-[(e)-1-(4-chlorophenyl)ethylideneamino]oxyacetate Chemical compound CN(C)CCOC(=O)CO\N=C(/C)C1=CC=C(Cl)C=C1 TXUGZLRUFAAHAO-LFIBNONCSA-N 0.000 description 1
- BMDZNGISRKXXIF-UHFFFAOYSA-N 2-(dimethylamino)ethyl 3-amino-4-chlorobenzoate Chemical compound CN(C)CCOC(=O)C1=CC=C(Cl)C(N)=C1 BMDZNGISRKXXIF-UHFFFAOYSA-N 0.000 description 1
- NYRSQWAKUMYFLI-UHFFFAOYSA-N 2-(furan-2-yl)-7-methyl-3,6-dihydroimidazo[4,5-f]quinolin-9-one Chemical compound N1C2=CC=C3NC(C)=CC(=O)C3=C2N=C1C1=CC=CO1 NYRSQWAKUMYFLI-UHFFFAOYSA-N 0.000 description 1
- BHANCCMWYDZQOR-UHFFFAOYSA-N 2-(methyldisulfanyl)pyridine Chemical compound CSSC1=CC=CC=N1 BHANCCMWYDZQOR-UHFFFAOYSA-N 0.000 description 1
- XTJMTDZHCLBKFU-UHFFFAOYSA-N 2-(tert-butylamino)-1-(2-fluorophenyl)ethanol Chemical compound CC(C)(C)NCC(O)C1=CC=CC=C1F XTJMTDZHCLBKFU-UHFFFAOYSA-N 0.000 description 1
- HCUWPRQGBLLKAF-UHFFFAOYSA-N 2-(tert-butylamino)-9,10-dimethoxy-6,7-dihydropyrimido[6,1-a]isoquinolin-4-one Chemical compound C1=2C=C(OC)C(OC)=CC=2CCN2C1=CC(NC(C)(C)C)=NC2=O HCUWPRQGBLLKAF-UHFFFAOYSA-N 0.000 description 1
- RBMHUYBJIYNRLY-UHFFFAOYSA-N 2-[(1-carboxy-1-hydroxyethyl)-hydroxyphosphoryl]-2-hydroxypropanoic acid Chemical compound OC(=O)C(O)(C)P(O)(=O)C(C)(O)C(O)=O RBMHUYBJIYNRLY-UHFFFAOYSA-N 0.000 description 1
- KSMAGQUYOIHWFS-ZETCQYMHSA-N 2-[(1s)-1-(2,6-dichlorophenoxy)ethyl]-4,5-dihydro-1h-imidazole Chemical compound O([C@@H](C)C=1NCCN=1)C1=C(Cl)C=CC=C1Cl KSMAGQUYOIHWFS-ZETCQYMHSA-N 0.000 description 1
- HQVZOORKDNCGCK-UHFFFAOYSA-N 2-[(2,4-dichlorophenyl)methyl]-4-(2,4,4-trimethylpentan-2-yl)phenol Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(O)C(CC=2C(=CC(Cl)=CC=2)Cl)=C1 HQVZOORKDNCGCK-UHFFFAOYSA-N 0.000 description 1
- LLECMGGNFBKPRH-UHFFFAOYSA-N 2-[(2,6-dimethylphenyl)carbamoyl]benzoic acid Chemical compound CC1=CC=CC(C)=C1NC(=O)C1=CC=CC=C1C(O)=O LLECMGGNFBKPRH-UHFFFAOYSA-N 0.000 description 1
- OKQHSIGMOWQUIK-UHFFFAOYSA-N 2-[(2-aminopurin-9-yl)methoxy]ethanol Chemical compound NC1=NC=C2N=CN(COCCO)C2=N1 OKQHSIGMOWQUIK-UHFFFAOYSA-N 0.000 description 1
- QZRIKRJFJVVUSF-UHFFFAOYSA-N 2-[(2-ethyl-1-benzofuran-3-yl)methyl]-4,5-dihydro-1h-imidazole Chemical compound CCC=1OC2=CC=CC=C2C=1CC1=NCCN1 QZRIKRJFJVVUSF-UHFFFAOYSA-N 0.000 description 1
- OGNRRAFRDFGFKA-UHFFFAOYSA-N 2-[(2-tert-butylphenyl)-phenylmethoxy]-n,n-dimethylethanamine Chemical compound C=1C=CC=C(C(C)(C)C)C=1C(OCCN(C)C)C1=CC=CC=C1 OGNRRAFRDFGFKA-UHFFFAOYSA-N 0.000 description 1
- IVSWFCVHTWYKEM-UHFFFAOYSA-N 2-[(3,6-dimethoxy-2,4-dimethylphenyl)methyl]-4,5-dihydro-1h-imidazole Chemical compound COC1=CC(C)=C(OC)C(C)=C1CC1=NCCN1 IVSWFCVHTWYKEM-UHFFFAOYSA-N 0.000 description 1
- XNMYNYSCEJBRPZ-UHFFFAOYSA-N 2-[(3-butyl-1-isoquinolinyl)oxy]-N,N-dimethylethanamine Chemical compound C1=CC=C2C(OCCN(C)C)=NC(CCCC)=CC2=C1 XNMYNYSCEJBRPZ-UHFFFAOYSA-N 0.000 description 1
- GPPLXTFJYCQMRB-UHFFFAOYSA-N 2-[(4-chloronaphthalen-1-yl)methyl]-4,5-dihydro-1h-imidazole Chemical compound C12=CC=CC=C2C(Cl)=CC=C1CC1=NCCN1 GPPLXTFJYCQMRB-UHFFFAOYSA-N 0.000 description 1
- HZNVLDCWAVIXRK-UHFFFAOYSA-N 2-[(4-chlorophenyl)-(1-methylpiperidin-4-ylidene)methyl]pyridine Chemical compound C1CN(C)CCC1=C(C=1N=CC=CC=1)C1=CC=C(Cl)C=C1 HZNVLDCWAVIXRK-UHFFFAOYSA-N 0.000 description 1
- YWQUFKJVMWHDSN-UHFFFAOYSA-N 2-[(4-ethylsulfanyl-3-methylpyridin-2-yl)methylsulfinyl]-1h-benzimidazole Chemical compound CCSC1=CC=NC(CS(=O)C=2NC3=CC=CC=C3N=2)=C1C YWQUFKJVMWHDSN-UHFFFAOYSA-N 0.000 description 1
- LBMFWYCMCHRLBU-SGIREYDYSA-N 2-[(6ar,9r,10ar)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline-9-yl]acetonitrile Chemical compound C1=CC([C@H]2C[C@H](CC#N)CN([C@@H]2C2)C)=C3C2=CNC3=C1 LBMFWYCMCHRLBU-SGIREYDYSA-N 0.000 description 1
- VHEJZMZHDUPRNV-UZYVYHOESA-N 2-[(z)-[(2,3-dichloro-4-methoxyphenyl)-(furan-2-yl)methylidene]amino]oxy-n,n-diethylethanamine Chemical compound C=1C=C(OC)C(Cl)=C(Cl)C=1C(=N/OCCN(CC)CC)/C1=CC=CO1 VHEJZMZHDUPRNV-UZYVYHOESA-N 0.000 description 1
- IKFQEQVEOQNTRJ-UHFFFAOYSA-N 2-[1-(4-chlorophenyl)-1-phenylethoxy]-n,n-diethylethanamine Chemical compound C=1C=C(Cl)C=CC=1C(C)(OCCN(CC)CC)C1=CC=CC=C1 IKFQEQVEOQNTRJ-UHFFFAOYSA-N 0.000 description 1
- BOCNKEKVCTZYIX-UHFFFAOYSA-M 2-[1-(5-bicyclo[2.2.1]hept-2-enyl)-1-phenylethoxy]ethyl-diethyl-methylazanium;bromide Chemical compound [Br-].C1C(C=C2)CC2C1C(C)(OCC[N+](C)(CC)CC)C1=CC=CC=C1 BOCNKEKVCTZYIX-UHFFFAOYSA-M 0.000 description 1
- XHNPXDMSCKEWJZ-UHFFFAOYSA-N 2-[1-[3-[2-(trifluoromethyl)phenothiazin-10-yl]propyl]piperidin-4-yl]oxyethanol Chemical compound C1CC(OCCO)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 XHNPXDMSCKEWJZ-UHFFFAOYSA-N 0.000 description 1
- YDDRXDOPBABLRB-UHFFFAOYSA-N 2-[2-(2,6-dichloroanilino)ethyl]guanidine Chemical compound NC(N)=NCCNC1=C(Cl)C=CC=C1Cl YDDRXDOPBABLRB-UHFFFAOYSA-N 0.000 description 1
- JBJASTVVFKIZBG-UHFFFAOYSA-N 2-[2-(2-methylpropyl)-4,5-diphenylpyrazol-3-yl]acetic acid Chemical compound OC(=O)CC=1N(CC(C)C)N=C(C=2C=CC=CC=2)C=1C1=CC=CC=C1 JBJASTVVFKIZBG-UHFFFAOYSA-N 0.000 description 1
- OXBBIHZWNDPBMQ-UHFFFAOYSA-N 2-[2-(4-benzhydrylpiperazin-1-yl)ethoxy]ethanol Chemical compound C1CN(CCOCCO)CCN1C(C=1C=CC=CC=1)C1=CC=CC=C1 OXBBIHZWNDPBMQ-UHFFFAOYSA-N 0.000 description 1
- OQKHPZHGTCHGOQ-UHFFFAOYSA-N 2-[2-(4-fluorophenyl)-5-nitroimidazol-1-yl]ethanol Chemical compound OCCN1C([N+]([O-])=O)=CN=C1C1=CC=C(F)C=C1 OQKHPZHGTCHGOQ-UHFFFAOYSA-N 0.000 description 1
- GFFFJSWQOUVZCY-UHFFFAOYSA-N 2-[2-(diethylamino)ethyl-ethylamino]ethyl 4-aminobenzoate Chemical compound CCN(CC)CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 GFFFJSWQOUVZCY-UHFFFAOYSA-N 0.000 description 1
- UCICRVXYPSKKJK-UHFFFAOYSA-N 2-[2-(diethylamino)ethyl]-5,11-dimethyl-6h-pyrido[4,3-b]carbazol-2-ium-9-ol;chloride Chemical compound [Cl-].C1=C(O)C=C2C3=C(C)C4=C[N+](CCN(CC)CC)=CC=C4C(C)=C3NC2=C1 UCICRVXYPSKKJK-UHFFFAOYSA-N 0.000 description 1
- RYOOHIUJEJZCFT-UHFFFAOYSA-N 2-[2-(diethylamino)ethylamino]-2-phenylacetic acid 3-methylbutyl ester Chemical compound CCN(CC)CCNC(C(=O)OCCC(C)C)C1=CC=CC=C1 RYOOHIUJEJZCFT-UHFFFAOYSA-N 0.000 description 1
- TVQRUKZAWSJVRX-UHFFFAOYSA-N 2-[2-[(3,4-dichlorobenzoyl)amino]phenoxy]acetic acid Chemical compound OC(=O)COC1=CC=CC=C1NC(=O)C1=CC=C(Cl)C(Cl)=C1 TVQRUKZAWSJVRX-UHFFFAOYSA-N 0.000 description 1
- CONZBLOLFWAHKJ-UHFFFAOYSA-L 2-[2-[carboxylatomethyl(carboxymethyl)amino]ethyl-(carboxymethyl)amino]acetate;cobalt;cobalt(2+) Chemical compound [Co].[Co+2].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O CONZBLOLFWAHKJ-UHFFFAOYSA-L 0.000 description 1
- FBMYKMYQHCBIGU-UHFFFAOYSA-N 2-[2-hydroxy-3-[[1-(1h-indol-3-yl)-2-methylpropan-2-yl]amino]propoxy]benzonitrile Chemical compound C=1NC2=CC=CC=C2C=1CC(C)(C)NCC(O)COC1=CC=CC=C1C#N FBMYKMYQHCBIGU-UHFFFAOYSA-N 0.000 description 1
- WWAZSAGOPXLSMS-UHFFFAOYSA-N 2-[3-(1h-imidazol-5-yl)propyl]-1-[2-(pyridin-2-ylamino)ethyl]guanidine Chemical compound C=1N=CNC=1CCCN=C(N)NCCNC1=CC=CC=N1 WWAZSAGOPXLSMS-UHFFFAOYSA-N 0.000 description 1
- XILVEPYQJIOVNB-UHFFFAOYSA-N 2-[3-(trifluoromethyl)anilino]benzoic acid 2-(2-hydroxyethoxy)ethyl ester Chemical compound OCCOCCOC(=O)C1=CC=CC=C1NC1=CC=CC(C(F)(F)F)=C1 XILVEPYQJIOVNB-UHFFFAOYSA-N 0.000 description 1
- GXPYCYWPUGKQIJ-UHFFFAOYSA-N 2-[3-(trifluoromethyl)phenyl]morpholine Chemical compound FC(F)(F)C1=CC=CC(C2OCCNC2)=C1 GXPYCYWPUGKQIJ-UHFFFAOYSA-N 0.000 description 1
- JWPMAPBNVNRWBX-UHFFFAOYSA-M 2-[3-[2-(2,4-dichlorophenyl)-2-[(2,4-dichlorophenyl)methoxy]ethyl]imidazol-1-ium-1-yl]-1-(4-fluorophenyl)ethanone;chloride Chemical compound [Cl-].C1=CC(F)=CC=C1C(=O)CN1C=[N+](CC(OCC=2C(=CC(Cl)=CC=2)Cl)C=2C(=CC(Cl)=CC=2)Cl)C=C1 JWPMAPBNVNRWBX-UHFFFAOYSA-M 0.000 description 1
- BGCVNLXQFOHWMO-UHFFFAOYSA-N 2-[3-[2-(3-methoxyphenyl)ethyl-methylamino]propyl]-2-(3,4,5-trimethoxyphenyl)tetradecanenitrile Chemical compound C=1C(OC)=C(OC)C(OC)=CC=1C(CCCCCCCCCCCC)(C#N)CCCN(C)CCC1=CC=CC(OC)=C1 BGCVNLXQFOHWMO-UHFFFAOYSA-N 0.000 description 1
- NLGUJWNOGYWZBI-UHFFFAOYSA-N 2-[3-chloro-4-(thiophene-2-carbonyl)phenyl]propanoic acid Chemical compound ClC1=CC(C(C(O)=O)C)=CC=C1C(=O)C1=CC=CS1 NLGUJWNOGYWZBI-UHFFFAOYSA-N 0.000 description 1
- AELILMBZWCGOSB-UHFFFAOYSA-N 2-[4-(2,6-dichloroanilino)thiophen-3-yl]acetic acid Chemical compound OC(=O)CC1=CSC=C1NC1=C(Cl)C=CC=C1Cl AELILMBZWCGOSB-UHFFFAOYSA-N 0.000 description 1
- TYCOFFBAZNSQOJ-UHFFFAOYSA-N 2-[4-(3-fluorophenyl)phenyl]propanoic acid Chemical compound C1=CC(C(C(O)=O)C)=CC=C1C1=CC=CC(F)=C1 TYCOFFBAZNSQOJ-UHFFFAOYSA-N 0.000 description 1
- JGQKYWPSFPCKLW-UHFFFAOYSA-N 2-[4-(4-bromophenyl)-1,3-thiazol-2-yl]propanoic acid Chemical compound S1C(C(C(O)=O)C)=NC(C=2C=CC(Br)=CC=2)=C1 JGQKYWPSFPCKLW-UHFFFAOYSA-N 0.000 description 1
- JIEKMACRVQTPRC-UHFFFAOYSA-N 2-[4-(4-chlorophenyl)-2-phenyl-5-thiazolyl]acetic acid Chemical compound OC(=O)CC=1SC(C=2C=CC=CC=2)=NC=1C1=CC=C(Cl)C=C1 JIEKMACRVQTPRC-UHFFFAOYSA-N 0.000 description 1
- DGRHWUCYLVPQJY-UHFFFAOYSA-N 2-[4-(trifluoromethyl)phenyl]indene-1,3-dione Chemical compound C1=CC(C(F)(F)F)=CC=C1C1C(=O)C2=CC=CC=C2C1=O DGRHWUCYLVPQJY-UHFFFAOYSA-N 0.000 description 1
- XFICNUNWUREFDP-UHFFFAOYSA-N 2-[4-[(7-chloroquinolin-4-yl)amino]pentylamino]ethanol Chemical compound ClC1=CC=C2C(NC(CCCNCCO)C)=CC=NC2=C1 XFICNUNWUREFDP-UHFFFAOYSA-N 0.000 description 1
- XDCDHIJFRRLVST-SNAWJCMRSA-N 2-[4-[(e)-3-(3,4,5-trimethoxyphenyl)prop-2-enoyl]piperazin-1-yl]acetic acid Chemical compound COC1=C(OC)C(OC)=CC(\C=C\C(=O)N2CCN(CC(O)=O)CC2)=C1 XDCDHIJFRRLVST-SNAWJCMRSA-N 0.000 description 1
- PGHWCZITRQNIPM-QPLCGJKRSA-N 2-[4-[(z)-2-chloro-1,2-diphenylethenyl]phenoxy]-n,n-diethylethanamine oxide Chemical compound C1=CC(OCC[N+]([O-])(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(/Cl)C1=CC=CC=C1 PGHWCZITRQNIPM-QPLCGJKRSA-N 0.000 description 1
- XYCYZUNVTJSIML-UHFFFAOYSA-N 2-[4-[2-(2-phenylbutanoyloxy)ethyl]piperazin-1-yl]ethyl 2-phenylbutanoate Chemical compound C=1C=CC=CC=1C(CC)C(=O)OCCN(CC1)CCN1CCOC(=O)C(CC)C1=CC=CC=C1 XYCYZUNVTJSIML-UHFFFAOYSA-N 0.000 description 1
- IULOBWFWYDMECP-UHFFFAOYSA-N 2-[4-[2-[(4-chlorophenyl)sulfonylamino]ethyl]phenyl]acetic acid Chemical compound C1=CC(CC(=O)O)=CC=C1CCNS(=O)(=O)C1=CC=C(Cl)C=C1 IULOBWFWYDMECP-UHFFFAOYSA-N 0.000 description 1
- VBTNUSHFBRTASB-UHFFFAOYSA-N 2-[4-[3-(3-chloropyrido[3,2-b][1,4]benzothiazin-10-yl)propyl]piperazin-1-yl]ethanol Chemical compound C1CN(CCO)CCN1CCCN1C2=NC=C(Cl)C=C2SC2=CC=CC=C21 VBTNUSHFBRTASB-UHFFFAOYSA-N 0.000 description 1
- ROZHWGHNJMGUJH-UHFFFAOYSA-N 2-[4-[3-(trifluoromethyl)phenyl]piperazin-1-yl]ethyl 2-[4-(2-methylpropyl)phenyl]propanoate Chemical compound C1=CC(CC(C)C)=CC=C1C(C)C(=O)OCCN1CCN(C=2C=C(C=CC=2)C(F)(F)F)CC1 ROZHWGHNJMGUJH-UHFFFAOYSA-N 0.000 description 1
- HDPDWVAJUZMKRW-UHFFFAOYSA-N 2-[4-[3-[6-(trifluoromethyl)thieno[2,3-b][1,4]benzothiazin-4-yl]propyl]piperazin-1-yl]ethanol Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=C1C=CS2 HDPDWVAJUZMKRW-UHFFFAOYSA-N 0.000 description 1
- ACZKTJZXXSHIGF-UHFFFAOYSA-N 2-[5-(4-chlorophenyl)pentyl]oxirane-2-carboxylic acid Chemical compound C=1C=C(Cl)C=CC=1CCCCCC1(C(=O)O)CO1 ACZKTJZXXSHIGF-UHFFFAOYSA-N 0.000 description 1
- NSCOCGOFKMUTMW-UHFFFAOYSA-N 2-[6-[[amino-[[amino-(4-chloroanilino)methylidene]amino]methylidene]amino]hexyl]-1-[amino-(4-chloroanilino)methylidene]guanidine;(4-aminophenyl)phosphonic acid Chemical compound NC1=CC=C(P(O)(O)=O)C=C1.NC1=CC=C(P(O)(O)=O)C=C1.C=1C=C(Cl)C=CC=1NC(/N)=N/C(N)=NCCCCCCN=C(N)\N=C(/N)NC1=CC=C(Cl)C=C1 NSCOCGOFKMUTMW-UHFFFAOYSA-N 0.000 description 1
- NWNBRKHCXIQLDT-UHFFFAOYSA-N 2-[6-methoxy-3-(4-methoxybenzoyl)-2-methylindol-1-yl]acetic acid Chemical compound C1=CC(OC)=CC=C1C(=O)C1=C(C)N(CC(O)=O)C2=CC(OC)=CC=C12 NWNBRKHCXIQLDT-UHFFFAOYSA-N 0.000 description 1
- LRYZPFWEZHSTHD-HEFFAWAOSA-O 2-[[(e,2s,3r)-2-formamido-3-hydroxyoctadec-4-enoxy]-hydroxyphosphoryl]oxyethyl-trimethylazanium Chemical class CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](NC=O)COP(O)(=O)OCC[N+](C)(C)C LRYZPFWEZHSTHD-HEFFAWAOSA-O 0.000 description 1
- XSYSSUAGVNOMCE-UHFFFAOYSA-N 2-[[2-(4-butylanilino)-2-oxoethyl]-(carboxymethyl)amino]acetic acid Chemical compound CCCCC1=CC=C(NC(=O)CN(CC(O)=O)CC(O)=O)C=C1 XSYSSUAGVNOMCE-UHFFFAOYSA-N 0.000 description 1
- VKPNJGFKWYNJJG-UHFFFAOYSA-N 2-[[4-(2,3-dihydro-1,4-benzodioxin-6-yl)-1,3-thiazol-2-yl]amino]-2-oxoacetic acid Chemical compound S1C(NC(=O)C(=O)O)=NC(C=2C=C3OCCOC3=CC=2)=C1 VKPNJGFKWYNJJG-UHFFFAOYSA-N 0.000 description 1
- PNAYGNCLPYBJAL-UHFFFAOYSA-N 2-[[4-chloro-6-(2,3-dimethylanilino)pyrimidin-2-yl]amino]acetic acid Chemical compound CC1=CC=CC(NC=2N=C(NCC(O)=O)N=C(Cl)C=2)=C1C PNAYGNCLPYBJAL-UHFFFAOYSA-N 0.000 description 1
- QWOLGQYXAQSNAE-UHFFFAOYSA-N 2-[[7-bromo-3-(2-fluorophenyl)-1,2-benzoxazol-6-yl]oxy]acetic acid Chemical compound N=1OC2=C(Br)C(OCC(=O)O)=CC=C2C=1C1=CC=CC=C1F QWOLGQYXAQSNAE-UHFFFAOYSA-N 0.000 description 1
- BYWMOMBWRFUAMC-UHFFFAOYSA-N 2-[bis(2-hydroxyethyl)amino]-n-[4-chloro-2-(2-chlorobenzoyl)phenyl]-n-methylacetamide Chemical compound OCCN(CCO)CC(=O)N(C)C1=CC=C(Cl)C=C1C(=O)C1=CC=CC=C1Cl BYWMOMBWRFUAMC-UHFFFAOYSA-N 0.000 description 1
- BBEDRGWUDRUNQC-UHFFFAOYSA-N 2-[bis(4-fluorophenyl)methoxy]ethanamine Chemical compound C=1C=C(F)C=CC=1C(OCCN)C1=CC=C(F)C=C1 BBEDRGWUDRUNQC-UHFFFAOYSA-N 0.000 description 1
- DVBXFRSRFUTZTN-UHFFFAOYSA-N 2-[carboxymethyl-[2-(2,6-diethyl-3-iodoanilino)-2-oxoethyl]amino]acetic acid Chemical compound CCC1=CC=C(I)C(CC)=C1NC(=O)CN(CC(O)=O)CC(O)=O DVBXFRSRFUTZTN-UHFFFAOYSA-N 0.000 description 1
- WNIDXAKKFOKNEF-UHFFFAOYSA-N 2-[carboxymethyl-[2-(2,6-diethylanilino)-2-oxoethyl]amino]acetic acid Chemical compound CCC1=CC=CC(CC)=C1NC(=O)CN(CC(O)=O)CC(O)=O WNIDXAKKFOKNEF-UHFFFAOYSA-N 0.000 description 1
- YMJMZFPZRVMNCH-FMIVXFBMSA-N 2-[methyl-[(e)-3-phenylprop-2-enyl]amino]-1-phenylpropan-1-ol Chemical compound C=1C=CC=CC=1/C=C/CN(C)C(C)C(O)C1=CC=CC=C1 YMJMZFPZRVMNCH-FMIVXFBMSA-N 0.000 description 1
- XKSAJZSJKURQRX-UHFFFAOYSA-N 2-acetyloxy-5-(4-fluorophenyl)benzoic acid Chemical compound C1=C(C(O)=O)C(OC(=O)C)=CC=C1C1=CC=C(F)C=C1 XKSAJZSJKURQRX-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- BUXRLJCGHZZYNE-UHFFFAOYSA-N 2-amino-5-[1-hydroxy-2-(propan-2-ylamino)ethyl]benzonitrile Chemical compound CC(C)NCC(O)C1=CC=C(N)C(C#N)=C1 BUXRLJCGHZZYNE-UHFFFAOYSA-N 0.000 description 1
- QOVUZUCXPAZXDZ-RXMQYKEDSA-N 2-amino-9-[(3r)-3,4-dihydroxybutyl]-3h-purin-6-one Chemical compound O=C1NC(N)=NC2=C1N=CN2CC[C@@H](O)CO QOVUZUCXPAZXDZ-RXMQYKEDSA-N 0.000 description 1
- MMPYYZUCLBHNSK-UHFFFAOYSA-N 2-amino-n-[4-(diethylamino)-2-methylphenyl]benzamide Chemical compound CC1=CC(N(CC)CC)=CC=C1NC(=O)C1=CC=CC=C1N MMPYYZUCLBHNSK-UHFFFAOYSA-N 0.000 description 1
- RYJTVDSLFJIKMP-STOWLHSFSA-N 2-benzhydryloxy-n,n-dimethylethanamine oxide;[(1r,4s)-7,7-dimethyl-3-oxo-4-bicyclo[2.2.1]heptanyl]methanesulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C.C=1C=CC=CC=1C(OCC[N+](C)([O-])C)C1=CC=CC=C1 RYJTVDSLFJIKMP-STOWLHSFSA-N 0.000 description 1
- SPCKHVPPRJWQRZ-UHFFFAOYSA-N 2-benzhydryloxy-n,n-dimethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 SPCKHVPPRJWQRZ-UHFFFAOYSA-N 0.000 description 1
- NCKMMSIFQUPKCK-UHFFFAOYSA-N 2-benzyl-4-chlorophenol Chemical compound OC1=CC=C(Cl)C=C1CC1=CC=CC=C1 NCKMMSIFQUPKCK-UHFFFAOYSA-N 0.000 description 1
- CMCCHHWTTBEZNM-UHFFFAOYSA-N 2-bromo-N-carbamoyl-3-methylbutanamide Chemical compound CC(C)C(Br)C(=O)NC(N)=O CMCCHHWTTBEZNM-UHFFFAOYSA-N 0.000 description 1
- IZQAUUVBKYXMET-UHFFFAOYSA-N 2-bromoethanamine Chemical compound NCCBr IZQAUUVBKYXMET-UHFFFAOYSA-N 0.000 description 1
- MNBLDUWBHRTWEP-UHFFFAOYSA-N 2-butoxyethyl 2-phenyl-2-piperidin-1-ylacetate Chemical compound C=1C=CC=CC=1C(C(=O)OCCOCCCC)N1CCCCC1 MNBLDUWBHRTWEP-UHFFFAOYSA-N 0.000 description 1
- KREZRKULMRIOKR-UHFFFAOYSA-N 2-butyl-1-(diaminomethylidene)-3-ethylguanidine Chemical compound CCCCN=C(NCC)N=C(N)N KREZRKULMRIOKR-UHFFFAOYSA-N 0.000 description 1
- QDGWHHFJDHIIOS-UHFFFAOYSA-N 2-chloro-1-(6-diethoxyphosphorylhexoxy)-4-methoxybenzene Chemical compound CCOP(=O)(OCC)CCCCCCOC1=CC=C(OC)C=C1Cl QDGWHHFJDHIIOS-UHFFFAOYSA-N 0.000 description 1
- PMPQRINMBYHVSP-MDZDMXLPSA-N 2-chloro-1-[(z)-n'-chlorocarbamimidoyl]iminoguanidine Chemical compound Cl/N=C(/N)\N=N\C(\N)=N\Cl PMPQRINMBYHVSP-MDZDMXLPSA-N 0.000 description 1
- JVVSVPLSTGMSJT-UHFFFAOYSA-N 2-chloro-1-[4-[4-(2-chloroacetyl)phenoxy]phenyl]ethanone Chemical compound C1=CC(C(=O)CCl)=CC=C1OC1=CC=C(C(=O)CCl)C=C1 JVVSVPLSTGMSJT-UHFFFAOYSA-N 0.000 description 1
- GXEUNRBWEAIPCN-UHFFFAOYSA-N 2-chloro-2-(3-chloro-4-cyclohexylphenyl)acetic acid Chemical compound ClC1=CC(C(Cl)C(=O)O)=CC=C1C1CCCCC1 GXEUNRBWEAIPCN-UHFFFAOYSA-N 0.000 description 1
- SYCYJNHKNPPDAT-UHFFFAOYSA-N 2-chloro-4-(1-hydroxyoctadecyl)benzoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)C1=CC=C(C(O)=O)C(Cl)=C1 SYCYJNHKNPPDAT-UHFFFAOYSA-N 0.000 description 1
- JIVPVXMEBJLZRO-CQSZACIVSA-N 2-chloro-5-[(1r)-1-hydroxy-3-oxo-2h-isoindol-1-yl]benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC([C@@]2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-CQSZACIVSA-N 0.000 description 1
- GGALEXMXDMUMDM-UHFFFAOYSA-N 2-chloro-6-(4-piperidinylthio)pyridine Chemical compound ClC1=CC=CC(SC2CCNCC2)=N1 GGALEXMXDMUMDM-UHFFFAOYSA-N 0.000 description 1
- KZMAWJRXKGLWGS-UHFFFAOYSA-N 2-chloro-n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]-n-(3-methoxypropyl)acetamide Chemical compound S1C(N(C(=O)CCl)CCCOC)=NC(C=2C=CC(OC)=CC=2)=C1 KZMAWJRXKGLWGS-UHFFFAOYSA-N 0.000 description 1
- FMMYTRQXHORTCU-UHFFFAOYSA-N 2-chloroethyl methanesulfonate Chemical compound CS(=O)(=O)OCCCl FMMYTRQXHORTCU-UHFFFAOYSA-N 0.000 description 1
- IJHSMJDMDSGQBT-YHUDWFPJSA-N 2-cyano-n-[(e)-(1-hydroxy-4-oxidoquinoxalin-4-ium-2-ylidene)methyl]iminoacetamide Chemical compound C1=CC=C2N(O)\C(=C\N=NC(=O)CC#N)C=[N+]([O-])C2=C1 IJHSMJDMDSGQBT-YHUDWFPJSA-N 0.000 description 1
- HPHBOJANXDKUQD-UHFFFAOYSA-N 2-cyanoacetohydrazide Chemical compound NNC(=O)CC#N HPHBOJANXDKUQD-UHFFFAOYSA-N 0.000 description 1
- AGJBLWCLQCKRJP-UHFFFAOYSA-N 2-cyclohexyl-2-phenylacetic acid 2-(diethylamino)ethyl ester Chemical compound C=1C=CC=CC=1C(C(=O)OCCN(CC)CC)C1CCCCC1 AGJBLWCLQCKRJP-UHFFFAOYSA-N 0.000 description 1
- MOUDDNPBPJHZHZ-UHFFFAOYSA-N 2-diazo-N-(2-hydrazinyl-2-oxoethyl)acetamide Chemical compound NNC(=O)CNC(=O)C=[N+]=[N-] MOUDDNPBPJHZHZ-UHFFFAOYSA-N 0.000 description 1
- GTQFGAJYMBBUSO-UHFFFAOYSA-N 2-diazoacetamide Chemical class NC(=O)C=[N+]=[N-] GTQFGAJYMBBUSO-UHFFFAOYSA-N 0.000 description 1
- PMHFLBQRALFMRT-UHFFFAOYSA-N 2-diphenylphosphorylacetohydrazide Chemical compound C=1C=CC=CC=1P(=O)(CC(=O)NN)C1=CC=CC=C1 PMHFLBQRALFMRT-UHFFFAOYSA-N 0.000 description 1
- HNJUBRBHULQQFX-UHFFFAOYSA-N 2-ethoxyethyl 5-chloro-1,3-benzoxazole-2-carboxylate Chemical compound ClC1=CC=C2OC(C(=O)OCCOCC)=NC2=C1 HNJUBRBHULQQFX-UHFFFAOYSA-N 0.000 description 1
- WVCHIGAIXREVNS-UHFFFAOYSA-N 2-hydroxy-1,4-naphthoquinone Chemical compound C1=CC=C2C(O)=CC(=O)C(=O)C2=C1 WVCHIGAIXREVNS-UHFFFAOYSA-N 0.000 description 1
- IVQOFBKHQCTVQV-UHFFFAOYSA-N 2-hydroxy-2,2-diphenylacetic acid 2-(diethylamino)ethyl ester Chemical compound C=1C=CC=CC=1C(O)(C(=O)OCCN(CC)CC)C1=CC=CC=C1 IVQOFBKHQCTVQV-UHFFFAOYSA-N 0.000 description 1
- QSAVEGSLJISCDF-UHFFFAOYSA-N 2-hydroxy-2-phenylacetic acid (1,2,2,6-tetramethyl-4-piperidinyl) ester Chemical compound C1C(C)(C)N(C)C(C)CC1OC(=O)C(O)C1=CC=CC=C1 QSAVEGSLJISCDF-UHFFFAOYSA-N 0.000 description 1
- CRLWISMVRNPGKN-UHFFFAOYSA-N 2-hydroxy-5-[(4-hydroxy-2-methyl-5-sulfophenyl)methyl]-4-methylbenzenesulfonic acid Chemical compound CC1=CC(O)=C(S(O)(=O)=O)C=C1CC1=CC(S(O)(=O)=O)=C(O)C=C1C CRLWISMVRNPGKN-UHFFFAOYSA-N 0.000 description 1
- UAZAHFDNEPPZHN-UHFFFAOYSA-M 2-hydroxy-n-methyl-n-[2-(1-methylpyrrolidin-1-ium-1-yl)ethyl]-2-phenyl-2-thiophen-2-ylacetamide;bromide Chemical compound [Br-].C=1C=CSC=1C(O)(C=1C=CC=CC=1)C(=O)N(C)CC[N+]1(C)CCCC1 UAZAHFDNEPPZHN-UHFFFAOYSA-M 0.000 description 1
- 239000001763 2-hydroxyethyl(trimethyl)azanium Substances 0.000 description 1
- IAWIJHCUEPVIOO-UHFFFAOYSA-N 2-imidazol-1-yl-1-[4-(2-phenylethyl)phenyl]ethanol Chemical compound C=1C=C(CCC=2C=CC=CC=2)C=CC=1C(O)CN1C=CN=C1 IAWIJHCUEPVIOO-UHFFFAOYSA-N 0.000 description 1
- CILDGVODBJAMGO-UHFFFAOYSA-N 2-methyl-1-(1-phenylethyl)imidazole Chemical compound C1=CN=C(C)N1C(C)C1=CC=CC=C1 CILDGVODBJAMGO-UHFFFAOYSA-N 0.000 description 1
- YWDWYOALXURQPZ-CYBMUJFWSA-N 2-methyl-n-[3-[(6s)-2,3,5,6-tetrahydroimidazo[2,1-b][1,3]thiazol-6-yl]phenyl]propanamide Chemical compound CC(C)C(=O)NC1=CC=CC([C@@H]2N=C3SCCN3C2)=C1 YWDWYOALXURQPZ-CYBMUJFWSA-N 0.000 description 1
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical group CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 1
- DHAXFWAIZHJURV-UHFFFAOYSA-N 2-phenoxyethyl 1-(3-cyano-3,3-diphenylpropyl)-4-phenylpiperidine-4-carboxylate;hydrochloride Chemical compound Cl.C1CN(CCC(C#N)(C=2C=CC=CC=2)C=2C=CC=CC=2)CCC1(C=1C=CC=CC=1)C(=O)OCCOC1=CC=CC=C1 DHAXFWAIZHJURV-UHFFFAOYSA-N 0.000 description 1
- YTRMTPPVNRALON-UHFFFAOYSA-N 2-phenyl-4-quinolinecarboxylic acid Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=CC=C1 YTRMTPPVNRALON-UHFFFAOYSA-N 0.000 description 1
- XOJUWOHYUFBSKE-UHFFFAOYSA-N 2-phosphonooxy-4-(trifluoromethyl)benzoic acid Chemical compound OC(=O)C1=CC=C(C(F)(F)F)C=C1OP(O)(O)=O XOJUWOHYUFBSKE-UHFFFAOYSA-N 0.000 description 1
- FFKUDWZICMJVPA-UHFFFAOYSA-N 2-phosphonooxybenzoic acid Chemical compound OC(=O)C1=CC=CC=C1OP(O)(O)=O FFKUDWZICMJVPA-UHFFFAOYSA-N 0.000 description 1
- VTBDTEWGXPOCSP-UHFFFAOYSA-N 2-piperidin-1-ylethyl 2-acetyloxy-2-benzyl-3-phenylpropanoate Chemical compound C=1C=CC=CC=1CC(C(=O)OCCN1CCCCC1)(OC(=O)C)CC1=CC=CC=C1 VTBDTEWGXPOCSP-UHFFFAOYSA-N 0.000 description 1
- DQOYUDHAZMAVLD-UHFFFAOYSA-N 2-pyrrolidin-1-ylethyl 2-[[7-(trifluoromethyl)quinolin-4-yl]amino]benzoate Chemical compound C=1C=NC2=CC(C(F)(F)F)=CC=C2C=1NC1=CC=CC=C1C(=O)OCCN1CCCC1 DQOYUDHAZMAVLD-UHFFFAOYSA-N 0.000 description 1
- IDMYDMZQNZFFBK-UHFFFAOYSA-N 2-pyrrolidin-1-ylethyl 4-butoxy-3,5-dimethoxybenzoate Chemical compound C1=C(OC)C(OCCCC)=C(OC)C=C1C(=O)OCCN1CCCC1 IDMYDMZQNZFFBK-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- JWUBBDSIWDLEOM-UHFFFAOYSA-N 25-Hydroxycholecalciferol Natural products C1CCC2(C)C(C(CCCC(C)(C)O)C)CCC2C1=CC=C1CC(O)CCC1=C JWUBBDSIWDLEOM-UHFFFAOYSA-N 0.000 description 1
- WELBEQNHUMMKEL-UHFFFAOYSA-N 3,3-diethyl-5-methyl-1h-pyridine-2,4-dione Chemical compound CCC1(CC)C(=O)NC=C(C)C1=O WELBEQNHUMMKEL-UHFFFAOYSA-N 0.000 description 1
- NMKSAYKQLCHXDK-UHFFFAOYSA-N 3,3-diphenyl-N-(1-phenylethyl)-1-propanamine Chemical compound C=1C=CC=CC=1C(C)NCCC(C=1C=CC=CC=1)C1=CC=CC=C1 NMKSAYKQLCHXDK-UHFFFAOYSA-N 0.000 description 1
- QFSBWEZVTZCUPC-UHFFFAOYSA-N 3,4,5-trimethoxy-n-(1-phenoxy-3-pyrrolidin-1-ylpropan-2-yl)benzamide Chemical compound COC1=C(OC)C(OC)=CC(C(=O)NC(COC=2C=CC=CC=2)CN2CCCC2)=C1 QFSBWEZVTZCUPC-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- HMKKFLSUPRUBOO-IUPFWZBJSA-N 3,4-dihydroxy-5-[3,4,5-tris[[(z)-octadec-9-enoyl]oxy]benzoyl]oxybenzoic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC1=C(OC(=O)CCCCCCC\C=C/CCCCCCCC)C(OC(=O)CCCCCCC\C=C/CCCCCCCC)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(O)=O)O)=C1 HMKKFLSUPRUBOO-IUPFWZBJSA-N 0.000 description 1
- INGVKLACODHWHI-UHFFFAOYSA-N 3,5-dibromo-2-hydroxy-n-(oxolan-2-ylmethyl)benzamide Chemical compound OC1=C(Br)C=C(Br)C=C1C(=O)NCC1OCCC1 INGVKLACODHWHI-UHFFFAOYSA-N 0.000 description 1
- SHZKJIKDCJBWRD-UHFFFAOYSA-N 3,5-dibromo-n-[(4-bromophenyl)methyl]-2-hydroxybenzamide Chemical compound OC1=C(Br)C=C(Br)C=C1C(=O)NCC1=CC=C(Br)C=C1 SHZKJIKDCJBWRD-UHFFFAOYSA-N 0.000 description 1
- KJBUBVMOTCYBPX-UHFFFAOYSA-N 3,5-dichloro-n-[2-(diethylamino)ethyl]-2-methoxybenzamide Chemical compound CCN(CC)CCNC(=O)C1=CC(Cl)=CC(Cl)=C1OC KJBUBVMOTCYBPX-UHFFFAOYSA-N 0.000 description 1
- ZPRPJXHUPKXCTE-UHFFFAOYSA-N 3,5-dimethyl-1h-pyridazin-6-one Chemical compound CC=1C=C(C)C(=O)NN=1 ZPRPJXHUPKXCTE-UHFFFAOYSA-N 0.000 description 1
- TUATYNXRYJTQTQ-BVRBKCERSA-N 3,6-diamino-n-[[(2s,5s,8z,11s,15s)-15-amino-11-(2-amino-1,4,5,6-tetrahydropyrimidin-6-yl)-8-[(carbamoylamino)methylidene]-2-(hydroxymethyl)-3,6,9,12,16-pentaoxo-1,4,7,10,13-pentazacyclohexadec-5-yl]methyl]hexanamide;3,6-diamino-n-[[(2s,5s,8z,11s,15s)-15-a Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.N1C(=O)\C(=C\NC(N)=O)NC(=O)[C@H](CNC(=O)CC(N)CCCN)NC(=O)[C@H](C)NC(=O)[C@@H](N)CNC(=O)[C@@H]1C1NC(=N)NCC1.N1C(=O)\C(=C\NC(N)=O)NC(=O)[C@H](CNC(=O)CC(N)CCCN)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CNC(=O)[C@@H]1C1NC(=N)NCC1 TUATYNXRYJTQTQ-BVRBKCERSA-N 0.000 description 1
- VPMZGRVNLHDREW-UHFFFAOYSA-N 3-(2-benzylindazol-3-yl)sulfanyl-n,n-dimethylpropan-1-amine Chemical compound N1=C2C=CC=CC2=C(SCCCN(C)C)N1CC1=CC=CC=C1 VPMZGRVNLHDREW-UHFFFAOYSA-N 0.000 description 1
- XLOYQLAMNLMXCE-UHFFFAOYSA-N 3-(2-ethoxyphenoxy)propane-1,2-diol Chemical compound CCOC1=CC=CC=C1OCC(O)CO XLOYQLAMNLMXCE-UHFFFAOYSA-N 0.000 description 1
- NUGLIYXAARVRPQ-UHFFFAOYSA-N 3-(2-nitroimidazol-1-yl)propane-1,2-diol Chemical compound OCC(O)CN1C=CN=C1[N+]([O-])=O NUGLIYXAARVRPQ-UHFFFAOYSA-N 0.000 description 1
- ZQTOZLYOIRKCPL-UHFFFAOYSA-N 3-(3,4,6,7,8,8a-hexahydro-1h-pyrrolo[1,2-a]pyrazin-2-yl)-1-(2-chlorophenothiazin-10-yl)propan-1-one Chemical compound C1CN2CCCC2CN1CCC(=O)N1C2=CC=CC=C2SC2=CC=C(Cl)C=C21 ZQTOZLYOIRKCPL-UHFFFAOYSA-N 0.000 description 1
- HOZLLXONNGPAPJ-UHFFFAOYSA-N 3-(3,4,6,7,8,8a-hexahydro-1h-pyrrolo[1,2-a]pyrazin-2-yl)-1-[2-(trifluoromethyl)phenothiazin-10-yl]propan-1-one Chemical compound C1CN2CCCC2CN1CCC(=O)N1C2=CC=CC=C2SC2=CC=C(C(F)(F)F)C=C21 HOZLLXONNGPAPJ-UHFFFAOYSA-N 0.000 description 1
- ZJWVOYSMOZCLEV-UHFFFAOYSA-N 3-(3,4-dimethoxyphenyl)-4-(hydroxymethyl)-4,5-dihydro-1h-pyridazin-6-one Chemical compound C1=C(OC)C(OC)=CC=C1C1=NNC(=O)CC1CO ZJWVOYSMOZCLEV-UHFFFAOYSA-N 0.000 description 1
- UTVDIOAGFGTMTM-UHFFFAOYSA-N 3-(3-bromophenyl)pyrrolidine-2,5-dione Chemical compound BrC1=CC=CC(C2C(NC(=O)C2)=O)=C1 UTVDIOAGFGTMTM-UHFFFAOYSA-N 0.000 description 1
- ICBQXEWYZVQCFH-UHFFFAOYSA-N 3-(4-bromoanilino)-n,n-dimethylpropanamide Chemical compound CN(C)C(=O)CCNC1=CC=C(Br)C=C1 ICBQXEWYZVQCFH-UHFFFAOYSA-N 0.000 description 1
- DOUQJBPSTIKRPH-UHFFFAOYSA-N 3-(4-methylpiperazin-1-yl)-1-[2-(trifluoromethyl)phenothiazin-10-yl]propan-1-one Chemical compound C1CN(C)CCN1CCC(=O)N1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 DOUQJBPSTIKRPH-UHFFFAOYSA-N 0.000 description 1
- PTVWPYVOOKLBCG-UHFFFAOYSA-N 3-(4-phenyl-1-piperazinyl)propane-1,2-diol Chemical compound C1CN(CC(O)CO)CCN1C1=CC=CC=C1 PTVWPYVOOKLBCG-UHFFFAOYSA-N 0.000 description 1
- KROWHJNUJMVHNU-UHFFFAOYSA-M 3-(5-bromofuran-2-carbonyl)oxybutyl-diethyl-methylazanium;iodide Chemical compound [I-].CC[N+](C)(CC)CCC(C)OC(=O)C1=CC=C(Br)O1 KROWHJNUJMVHNU-UHFFFAOYSA-M 0.000 description 1
- MLLYDWHLZFTQBY-UHFFFAOYSA-N 3-(carboxymethylsulfanyl)propanoic acid Chemical compound OC(=O)CCSCC(O)=O MLLYDWHLZFTQBY-UHFFFAOYSA-N 0.000 description 1
- BDNMABJZSXTKAQ-VCPDFFEISA-N 3-(diethylamino)propyl (1r,4s)-3-phenylbicyclo[2.2.1]heptane-3-carboxylate Chemical compound CCN(CC)CCCOC(=O)C1([C@H]2CC[C@H](C2)C1)C1=CC=CC=C1 BDNMABJZSXTKAQ-VCPDFFEISA-N 0.000 description 1
- JMUAKWNHKQBPGJ-UHFFFAOYSA-N 3-(pyridin-2-yldisulfanyl)-n-[4-[3-(pyridin-2-yldisulfanyl)propanoylamino]butyl]propanamide Chemical compound C=1C=CC=NC=1SSCCC(=O)NCCCCNC(=O)CCSSC1=CC=CC=N1 JMUAKWNHKQBPGJ-UHFFFAOYSA-N 0.000 description 1
- TVZRAEYQIKYCPH-UHFFFAOYSA-N 3-(trimethylsilyl)propane-1-sulfonic acid Chemical compound C[Si](C)(C)CCCS(O)(=O)=O TVZRAEYQIKYCPH-UHFFFAOYSA-N 0.000 description 1
- SDOJJWYOCLRIRI-UHFFFAOYSA-N 3-[(2,2-diphenylcyclopropyl)methylamino]propyl 3,4,5-trimethoxybenzoate Chemical compound COC1=C(OC)C(OC)=CC(C(=O)OCCCNCC2C(C2)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 SDOJJWYOCLRIRI-UHFFFAOYSA-N 0.000 description 1
- IQBKPMNLEWNTCN-UHFFFAOYSA-N 3-[(4-chlorophenyl)methyl]-2,3,4,6,7,8,9,9a-octahydro-1h-quinolizine Chemical compound C1=CC(Cl)=CC=C1CC1CN2CCCCC2CC1 IQBKPMNLEWNTCN-UHFFFAOYSA-N 0.000 description 1
- SBHNNNRQZGYOAU-YVEFUNNKSA-N 3-[(6ar,9s)-5-bromo-7-methyl-6,6a,8,9-tetrahydro-4h-indolo[4,3-fg]quinoline-9-yl]-1,1-diethylurea Chemical compound C1=CC(C=2[C@H](N(C)C[C@H](C=2)NC(=O)N(CC)CC)C2)=C3C2=C(Br)NC3=C1 SBHNNNRQZGYOAU-YVEFUNNKSA-N 0.000 description 1
- LVMVXZOPCAMYHC-QOAXCGLXSA-N 3-[(6ar,9s,10ar)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline-9-yl]-2-cyanopropanamide Chemical compound C1=CC([C@H]2C[C@@H](CC(C#N)C(N)=O)CN([C@@H]2C2)C)=C3C2=CNC3=C1 LVMVXZOPCAMYHC-QOAXCGLXSA-N 0.000 description 1
- BURFYXOPVVMBNO-JZYPHBBUSA-N 3-[(e)-[(2e)-2-[(4-chlorophenyl)methylidene]cyclohexylidene]amino]oxy-n,n-di(propan-2-yl)propan-1-amine Chemical compound CC(C)N(C(C)C)CCCO\N=C\1/CCCC/C/1=C\C1=CC=C(Cl)C=C1 BURFYXOPVVMBNO-JZYPHBBUSA-N 0.000 description 1
- PRGQOVDEZVJQJK-UHFFFAOYSA-N 3-[1-[3-(2-chlorophenothiazin-10-yl)propyl]-4-hydroxypiperidin-4-yl]oxolan-2-one Chemical compound C1CN(CCCN2C3=CC(Cl)=CC=C3SC3=CC=CC=C32)CCC1(O)C1CCOC1=O PRGQOVDEZVJQJK-UHFFFAOYSA-N 0.000 description 1
- XEBSDNCEYSPGIU-UHFFFAOYSA-N 3-[1-[4-(2-chloroethyl)phenyl]butyl]-4-hydroxychromen-2-one Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CCC)C1=CC=C(CCCl)C=C1 XEBSDNCEYSPGIU-UHFFFAOYSA-N 0.000 description 1
- FEBOTPHFXYHVPL-UHFFFAOYSA-N 3-[1-[4-(4-fluorophenyl)-4-oxobutyl]-4-piperidinyl]-1H-benzimidazol-2-one Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCC(N2C(NC3=CC=CC=C32)=O)CC1 FEBOTPHFXYHVPL-UHFFFAOYSA-N 0.000 description 1
- KFKRXESVMDBTNQ-UHFFFAOYSA-N 3-[18-(2-carboxylatoethyl)-8,13-bis(1-hydroxyethyl)-3,7,12,17-tetramethyl-22,23-dihydroporphyrin-21,24-diium-2-yl]propanoate Chemical class N1C2=C(C)C(C(C)O)=C1C=C(N1)C(C)=C(C(O)C)C1=CC(C(C)=C1CCC(O)=O)=NC1=CC(C(CCC(O)=O)=C1C)=NC1=C2 KFKRXESVMDBTNQ-UHFFFAOYSA-N 0.000 description 1
- BXQFWCBDMKMQBG-UHFFFAOYSA-N 3-[2-[(4-chlorophenyl)-phenylmethoxy]ethyl]-1,3-oxazinane Chemical compound C1=CC(Cl)=CC=C1C(C=1C=CC=CC=1)OCCN1COCCC1 BXQFWCBDMKMQBG-UHFFFAOYSA-N 0.000 description 1
- QACGCDWSRFDWQO-UHFFFAOYSA-N 3-[2-[3-(tert-butylamino)-2-hydroxypropoxy]-4-chlorophenyl]cyclopent-2-en-1-one Chemical compound CC(C)(C)NCC(O)COC1=CC(Cl)=CC=C1C1=CC(=O)CC1 QACGCDWSRFDWQO-UHFFFAOYSA-N 0.000 description 1
- LNSHWIZFFHXFPQ-UHFFFAOYSA-N 3-[2-[4-[[3-(furan-2-ylmethyl)imidazo[4,5-b]pyridin-2-yl]amino]piperidin-1-yl]ethyl]-2-methylpyrido[1,2-a]pyrimidin-4-one Chemical compound CC=1N=C2C=CC=CN2C(=O)C=1CCN(CC1)CCC1NC1=NC2=CC=CN=C2N1CC1=CC=CO1 LNSHWIZFFHXFPQ-UHFFFAOYSA-N 0.000 description 1
- DEQLGSOHGTZKFB-UHFFFAOYSA-N 3-[3-(1h-imidazol-3-ium-3-ylmethyl)-2-methylindol-1-yl]propanoate Chemical compound C12=CC=CC=C2N(CCC(O)=O)C(C)=C1CN1C=CN=C1 DEQLGSOHGTZKFB-UHFFFAOYSA-N 0.000 description 1
- FXZJKVODWNYPKK-UHFFFAOYSA-N 3-[3-[4-(3-chlorophenyl)piperazin-1-yl]propyl]-1h-quinazoline-2,4-dione Chemical compound ClC1=CC=CC(N2CCN(CCCN3C(C4=CC=CC=C4NC3=O)=O)CC2)=C1 FXZJKVODWNYPKK-UHFFFAOYSA-N 0.000 description 1
- VNLMLHDKNUIWNM-UHFFFAOYSA-N 3-[3-[4-(4-fluorobenzoyl)piperidin-1-yl]propyl]-1h-benzimidazol-2-one Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCCN2C(NC3=CC=CC=C32)=O)CC1 VNLMLHDKNUIWNM-UHFFFAOYSA-N 0.000 description 1
- CNBMQDMRPDUMDK-UHFFFAOYSA-N 3-[3-[4-(5-chloro-2-oxo-3h-benzimidazol-1-yl)piperidin-1-yl]propyl]-6-fluoro-1h-benzimidazol-2-one Chemical compound C12=CC=C(Cl)C=C2NC(=O)N1C(CC1)CCN1CCCN1C2=CC=C(F)C=C2NC1=O CNBMQDMRPDUMDK-UHFFFAOYSA-N 0.000 description 1
- KRMKBOUUDPUIDK-UHFFFAOYSA-N 3-[4-(2-hydroxyethyl)-1-piperazinyl]-1-[2-(trifluoromethyl)-10-phenothiazinyl]-1-propanone Chemical compound C1CN(CCO)CCN1CCC(=O)N1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 KRMKBOUUDPUIDK-UHFFFAOYSA-N 0.000 description 1
- MBAQRSKEYXLLNY-UHFFFAOYSA-N 3-[4-(4-fluorophenyl)-3,6-dihydro-2h-pyridin-1-yl]-1-[1-(2-hydroxyethyl)-5-methylpyrazol-4-yl]propan-1-one Chemical compound C1=NN(CCO)C(C)=C1C(=O)CCN1CC=C(C=2C=CC(F)=CC=2)CC1 MBAQRSKEYXLLNY-UHFFFAOYSA-N 0.000 description 1
- WXFFTUKVTRHPLQ-UHFFFAOYSA-N 3-[4-[3-(2-chlorothioxanthen-9-ylidene)propyl]piperazin-1-yl]-n-methylpropanamide Chemical compound C1CN(CCC(=O)NC)CCN1CCC=C1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 WXFFTUKVTRHPLQ-UHFFFAOYSA-N 0.000 description 1
- MZELTXWHFDTAOO-UHFFFAOYSA-N 3-[4-[3-(tert-butylamino)-2-hydroxypropoxy]phenyl]-7-methoxy-2-methylisoquinolin-1-one Chemical compound CN1C(=O)C2=CC(OC)=CC=C2C=C1C1=CC=C(OCC(O)CNC(C)(C)C)C=C1 MZELTXWHFDTAOO-UHFFFAOYSA-N 0.000 description 1
- MLDQSYUQSLUEPG-UHFFFAOYSA-N 3-[4-[4-(4-fluorobenzoyl)piperidin-1-yl]butyl]-1h-quinazoline-2,4-dione Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCCCN2C(C3=CC=CC=C3NC2=O)=O)CC1 MLDQSYUQSLUEPG-UHFFFAOYSA-N 0.000 description 1
- PLZMRGRLCWCLFW-UHFFFAOYSA-N 3-[5-(3-bromophenyl)tetrazol-2-yl]-1-piperidin-1-ylpropan-1-one Chemical compound BrC1=CC=CC(C2=NN(CCC(=O)N3CCCCC3)N=N2)=C1 PLZMRGRLCWCLFW-UHFFFAOYSA-N 0.000 description 1
- MVVJINIUPYKZHR-UHFFFAOYSA-N 3-[[4-[5-(methoxymethyl)-2-oxo-1,3-oxazolidin-3-yl]phenoxy]methyl]benzonitrile Chemical compound O=C1OC(COC)CN1C(C=C1)=CC=C1OCC1=CC=CC(C#N)=C1 MVVJINIUPYKZHR-UHFFFAOYSA-N 0.000 description 1
- FQWNGSKQHPNIQG-UHFFFAOYSA-N 3-[[bis(2-chloroethyl)amino-(2-chloroethoxy)phosphoryl]amino]propan-1-ol Chemical compound OCCCNP(=O)(OCCCl)N(CCCl)CCCl FQWNGSKQHPNIQG-UHFFFAOYSA-N 0.000 description 1
- HGWOCGUIQUERSN-UHFFFAOYSA-N 3-acetamido-2,4,6-tribromo-5-(2-hydroxyethylcarbamoyl)benzoic acid Chemical compound CC(=O)NC1=C(Br)C(C(O)=O)=C(Br)C(C(=O)NCCO)=C1Br HGWOCGUIQUERSN-UHFFFAOYSA-N 0.000 description 1
- CUNJTOHTJOOFJQ-WZTVWXICSA-N 3-acetamido-2,4,6-triiodobenzoic acid;(2r,3r,4r,5s)-6-(methylamino)hexane-1,2,3,4,5-pentol Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CC(=O)NC1=C(I)C=C(I)C(C(O)=O)=C1I CUNJTOHTJOOFJQ-WZTVWXICSA-N 0.000 description 1
- CURYRIVJTBNEGU-UHFFFAOYSA-L 3-bromo-1-[12-(3-bromopropanoyl)-3,12-diaza-6,9-diazoniadispiro[5.2.5^{9}.2^{6}]hexadecan-3-yl]propan-1-one;dichloride Chemical compound [Cl-].[Cl-].C1CN(C(=O)CCBr)CC[N+]21CC[N+]1(CCN(CC1)C(=O)CCBr)CC2 CURYRIVJTBNEGU-UHFFFAOYSA-L 0.000 description 1
- CDOUZKKFHVEKRI-UHFFFAOYSA-N 3-bromo-n-[(prop-2-enoylamino)methyl]propanamide Chemical compound BrCCC(=O)NCNC(=O)C=C CDOUZKKFHVEKRI-UHFFFAOYSA-N 0.000 description 1
- IDQGRVRLINQKBK-UHFFFAOYSA-N 3-chloro-1-imidazol-1-yl-4-phenylisoquinoline Chemical compound C=12C=CC=CC2=C(C=2C=CC=CC=2)C(Cl)=NC=1N1C=CN=C1 IDQGRVRLINQKBK-UHFFFAOYSA-N 0.000 description 1
- BITWVVLBVUMFHU-UHFFFAOYSA-N 3-chloro-n-(2-phenylethyl)propanamide Chemical compound ClCCC(=O)NCCC1=CC=CC=C1 BITWVVLBVUMFHU-UHFFFAOYSA-N 0.000 description 1
- MFERNRNDCJFIAS-UHFFFAOYSA-N 3-cyclohexyl-3-hydroxybutanoic acid Chemical compound OC(=O)CC(O)(C)C1CCCCC1 MFERNRNDCJFIAS-UHFFFAOYSA-N 0.000 description 1
- ABQLAMJAQZFPJI-UHFFFAOYSA-N 3-heptyloxolan-2-one Chemical compound CCCCCCCC1CCOC1=O ABQLAMJAQZFPJI-UHFFFAOYSA-N 0.000 description 1
- HNPVERUJGFNNRV-UHFFFAOYSA-N 3-iodophthalic acid Chemical compound OC(=O)C1=CC=CC(I)=C1C(O)=O HNPVERUJGFNNRV-UHFFFAOYSA-N 0.000 description 1
- GINRFPGQEWNBJV-UHFFFAOYSA-N 3-methyl-2-(4,5,6,7-tetrahydro-1,3-benzothiazol-2-yl)-3,4-dihydropyrazol-5-amine Chemical compound CC1CC(N)=NN1C(S1)=NC2=C1CCCC2 GINRFPGQEWNBJV-UHFFFAOYSA-N 0.000 description 1
- HKVGEMSALZULPM-UHFFFAOYSA-N 3-methylbutyl 2-phenyl-2-(2-pyrrolidin-1-ylethylamino)acetate Chemical compound C=1C=CC=CC=1C(C(=O)OCCC(C)C)NCCN1CCCC1 HKVGEMSALZULPM-UHFFFAOYSA-N 0.000 description 1
- AYKYXWQEBUNJCN-UHFFFAOYSA-N 3-methylfuran-2,5-dione Chemical compound CC1=CC(=O)OC1=O AYKYXWQEBUNJCN-UHFFFAOYSA-N 0.000 description 1
- LINVVMHRTUSXHL-GGVPDPBRSA-N 3beta-hydroxy-5alpha-cholest-8(14)-en-15-one Chemical compound C([C@@H]1CC2)[C@@H](O)CC[C@]1(C)[C@@H]1C2=C2C(=O)C[C@H]([C@H](C)CCCC(C)C)[C@@]2(C)CC1 LINVVMHRTUSXHL-GGVPDPBRSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- UHBAPGWWRFVTFS-UHFFFAOYSA-N 4,4'-dipyridyl disulfide Chemical compound C=1C=NC=CC=1SSC1=CC=NC=C1 UHBAPGWWRFVTFS-UHFFFAOYSA-N 0.000 description 1
- DAIZVBCVNQSHLI-UHFFFAOYSA-N 4,4-diphenyl-1-(propan-2-ylamino)butan-2-ol Chemical compound C=1C=CC=CC=1C(CC(O)CNC(C)C)C1=CC=CC=C1 DAIZVBCVNQSHLI-UHFFFAOYSA-N 0.000 description 1
- UVKFSMBPRQBNCH-UHFFFAOYSA-M 4,4-diphenylbutan-2-yl-ethyl-dimethylazanium;bromide Chemical compound [Br-].C=1C=CC=CC=1C(CC(C)[N+](C)(C)CC)C1=CC=CC=C1 UVKFSMBPRQBNCH-UHFFFAOYSA-M 0.000 description 1
- TYNLGDBUJLVSMA-UHFFFAOYSA-N 4,5-diacetyloxy-9,10-dioxo-2-anthracenecarboxylic acid Chemical compound O=C1C2=CC(C(O)=O)=CC(OC(C)=O)=C2C(=O)C2=C1C=CC=C2OC(=O)C TYNLGDBUJLVSMA-UHFFFAOYSA-N 0.000 description 1
- PMAHPMMCPXYARU-UHFFFAOYSA-N 4-(1-methylpiperidin-1-ium-1-yl)-2,2-diphenylbutanamide;bromide Chemical compound [Br-].C=1C=CC=CC=1C(C(N)=O)(C=1C=CC=CC=1)CC[N+]1(C)CCCCC1 PMAHPMMCPXYARU-UHFFFAOYSA-N 0.000 description 1
- OWYLAEYXIQKAOL-UHFFFAOYSA-N 4-(1-pyrrolidinyl)-1-(2,4,6-trimethoxyphenyl)-1-butanone Chemical compound COC1=CC(OC)=CC(OC)=C1C(=O)CCCN1CCCC1 OWYLAEYXIQKAOL-UHFFFAOYSA-N 0.000 description 1
- CWEUKXMDWMAICX-UHFFFAOYSA-N 4-(2,6-dichloroanilino)thiophene-3-carboxylic acid Chemical compound OC(=O)C1=CSC=C1NC1=C(Cl)C=CC=C1Cl CWEUKXMDWMAICX-UHFFFAOYSA-N 0.000 description 1
- QUXRAOQDUJFWIA-UHFFFAOYSA-N 4-(2-chloroxanthen-9-ylidene)-1-methylpiperidine Chemical compound C1CN(C)CCC1=C1C2=CC(Cl)=CC=C2OC2=CC=CC=C21 QUXRAOQDUJFWIA-UHFFFAOYSA-N 0.000 description 1
- PQLYZHNINGGVMB-UHFFFAOYSA-N 4-(3,4,6,7,8,8a-hexahydro-1h-pyrrolo[1,2-a]pyrazin-2-yl)-1-(4-fluorophenyl)butan-1-one Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CC2CCCN2CC1 PQLYZHNINGGVMB-UHFFFAOYSA-N 0.000 description 1
- QPIRWBYRKYGHDQ-UHFFFAOYSA-N 4-(4-chlorophenyl)-5-[2-(4-phenylpiperazin-1-yl)ethyl]-1,3-dioxol-2-one Chemical compound C1=CC(Cl)=CC=C1C1=C(CCN2CCN(CC2)C=2C=CC=CC=2)OC(=O)O1 QPIRWBYRKYGHDQ-UHFFFAOYSA-N 0.000 description 1
- HBLQZEWZVJXHKQ-UHFFFAOYSA-M 4-(4-chlorophenyl)butyl-diethyl-heptylazanium;dihydrogen phosphate Chemical compound OP(O)([O-])=O.CCCCCCC[N+](CC)(CC)CCCCC1=CC=C(Cl)C=C1 HBLQZEWZVJXHKQ-UHFFFAOYSA-M 0.000 description 1
- NDPOGPAZKKPOPV-UHFFFAOYSA-N 4-(4-ethylphenyl)piperidine Chemical compound C1=CC(CC)=CC=C1C1CCNCC1 NDPOGPAZKKPOPV-UHFFFAOYSA-N 0.000 description 1
- NFYAWNPZCNDDOG-UHFFFAOYSA-N 4-(4-fluorophenyl)-5-[2-(4-phenylpiperazin-1-yl)ethyl]-1,3-dioxol-2-one Chemical compound C1=CC(F)=CC=C1C1=C(CCN2CCN(CC2)C=2C=CC=CC=2)OC(=O)O1 NFYAWNPZCNDDOG-UHFFFAOYSA-N 0.000 description 1
- MDCYBLVSLOPFAZ-UHFFFAOYSA-N 4-(6-imino-5-methyl-3-phenylpyridazin-1-yl)butanoic acid;hydrochloride Chemical compound Cl.OC(=O)CCCN1C(=N)C(C)=CC(C=2C=CC=CC=2)=N1 MDCYBLVSLOPFAZ-UHFFFAOYSA-N 0.000 description 1
- UPXRTVAIJMUAQR-UHFFFAOYSA-N 4-(9h-fluoren-9-ylmethoxycarbonylamino)-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-2-carboxylic acid Chemical compound C1C(C(O)=O)N(C(=O)OC(C)(C)C)CC1NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 UPXRTVAIJMUAQR-UHFFFAOYSA-N 0.000 description 1
- QXWKHSSBFQDQPR-UHFFFAOYSA-N 4-(hexadecylamino)benzoic acid Chemical compound CCCCCCCCCCCCCCCCNC1=CC=C(C(O)=O)C=C1 QXWKHSSBFQDQPR-UHFFFAOYSA-N 0.000 description 1
- AKJHMTWEGVYYSE-AIRMAKDCSA-N 4-HPR Chemical compound C=1C=C(O)C=CC=1NC(=O)/C=C(\C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C AKJHMTWEGVYYSE-AIRMAKDCSA-N 0.000 description 1
- MAUUNYHAXGTMMQ-AMXXQSRZSA-N 4-[(2e)-2-[(3as,4s,5r,6as)-5-hydroxy-4-[(3s,4s)-3-hydroxy-4-methylnona-1,6-diynyl]-3,3a,4,5,6,6a-hexahydro-1h-pentalen-2-ylidene]ethoxy]butanoic acid Chemical compound C1\C(=C/COCCCC(O)=O)C[C@@H]2[C@@H](C#C[C@@H](O)[C@@H](C)CC#CCC)[C@H](O)C[C@@H]21 MAUUNYHAXGTMMQ-AMXXQSRZSA-N 0.000 description 1
- AXDJCCTWPBKUKL-UHFFFAOYSA-N 4-[(4-aminophenyl)-(4-imino-3-methylcyclohexa-2,5-dien-1-ylidene)methyl]aniline;hydron;chloride Chemical compound Cl.C1=CC(=N)C(C)=CC1=C(C=1C=CC(N)=CC=1)C1=CC=C(N)C=C1 AXDJCCTWPBKUKL-UHFFFAOYSA-N 0.000 description 1
- KBSZKZDILXBWMX-UHFFFAOYSA-N 4-[(4-benzhydrylpiperazin-1-yl)methyl]phenol Chemical compound C1=CC(O)=CC=C1CN1CCN(C(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 KBSZKZDILXBWMX-UHFFFAOYSA-N 0.000 description 1
- KXSKBNFNSAMNEZ-UHFFFAOYSA-N 4-[(4-benzyl-6-methyl-1,3-dihydrofuro[3,4-c]pyridin-7-yl)oxy]-n-propan-2-ylbutan-1-amine Chemical compound N1=C(C)C(OCCCCNC(C)C)=C2COCC2=C1CC1=CC=CC=C1 KXSKBNFNSAMNEZ-UHFFFAOYSA-N 0.000 description 1
- MXEAHCWVILOHDC-UHFFFAOYSA-N 4-[(4-chlorophenyl)methyl]-6,6-dimethyl-1,7-dihydroimidazo[1,2-a]purin-9-one Chemical compound N=1C(C)(C)CN(C(C=2NC=NC=22)=O)C=1N2CC1=CC=C(Cl)C=C1 MXEAHCWVILOHDC-UHFFFAOYSA-N 0.000 description 1
- YNWCUCSDUMVJKR-UHFFFAOYSA-N 4-[(7-chloroquinolin-4-yl)amino]-2-(pyrrolidin-1-ylmethyl)phenol Chemical compound OC1=CC=C(NC=2C3=CC=C(Cl)C=C3N=CC=2)C=C1CN1CCCC1 YNWCUCSDUMVJKR-UHFFFAOYSA-N 0.000 description 1
- HXSSCTOWBWPFEV-XCVCLJGOSA-N 4-[(e)-3-[4-(carboxymethoxy)phenyl]-3-oxoprop-1-enyl]benzoic acid Chemical compound C1=CC(OCC(=O)O)=CC=C1C(=O)\C=C\C1=CC=C(C(O)=O)C=C1 HXSSCTOWBWPFEV-XCVCLJGOSA-N 0.000 description 1
- URIZBPYQIRFMBF-UHFFFAOYSA-N 4-[1-[3-methyl-5-(5-oxo-2h-furan-3-yl)-1-benzofuran-2-yl]ethoxy]-4-oxobutanoic acid Chemical compound C1=C2C(C)=C(C(OC(=O)CCC(O)=O)C)OC2=CC=C1C1=CC(=O)OC1 URIZBPYQIRFMBF-UHFFFAOYSA-N 0.000 description 1
- DDBFLXGTCAVAFD-UHFFFAOYSA-N 4-[1-hydroxy-2-(methylamino)propyl]benzene-1,2-diol Chemical compound CNC(C)C(O)C1=CC=C(O)C(O)=C1 DDBFLXGTCAVAFD-UHFFFAOYSA-N 0.000 description 1
- MPCPSVWSWKWJLO-UHFFFAOYSA-N 4-[1-hydroxy-2-(propan-2-ylamino)ethyl]phenol Chemical compound CC(C)NCC(O)C1=CC=C(O)C=C1 MPCPSVWSWKWJLO-UHFFFAOYSA-N 0.000 description 1
- FZPBXNLCWHEGFO-UHFFFAOYSA-N 4-[2-(4-carboxyphenoxy)propan-2-yloxy]benzoic acid Chemical compound C(=O)(O)C1=CC=C(OC(C)(C)OC2=CC=C(C=C2)C(=O)O)C=C1 FZPBXNLCWHEGFO-UHFFFAOYSA-N 0.000 description 1
- IEZSLVKNOOIGQP-UHFFFAOYSA-N 4-[2-(6-chloropyridin-2-yl)sulfanylethyl]morpholine Chemical compound ClC1=CC=CC(SCCN2CCOCC2)=N1 IEZSLVKNOOIGQP-UHFFFAOYSA-N 0.000 description 1
- BMMHZTIQZODVHZ-UHFFFAOYSA-N 4-[2-[[2-[3-amino-5-(hydroxymethyl)phenyl]-2-hydroxyethyl]amino]propyl]phenol Chemical compound C=1C(N)=CC(CO)=CC=1C(O)CNC(C)CC1=CC=C(O)C=C1 BMMHZTIQZODVHZ-UHFFFAOYSA-N 0.000 description 1
- AQMBNKMVOOFSOJ-UHFFFAOYSA-N 4-[2-hydroxy-3-[4-oxo-2-(2h-tetrazol-5-yl)chromen-5-yl]oxypropoxy]benzonitrile Chemical compound C=1C=CC=2OC(C3=NNN=N3)=CC(=O)C=2C=1OCC(O)COC1=CC=C(C#N)C=C1 AQMBNKMVOOFSOJ-UHFFFAOYSA-N 0.000 description 1
- UXHRUDKGYGPMIE-UHFFFAOYSA-N 4-[3-(2-chloro-4-phenylphenoxy)propyl]morpholine Chemical compound ClC1=CC(C=2C=CC=CC=2)=CC=C1OCCCN1CCOCC1 UXHRUDKGYGPMIE-UHFFFAOYSA-N 0.000 description 1
- SJYFDORQYYEJLB-UHFFFAOYSA-N 4-[3-(tert-butylamino)-2-hydroxypropoxy]-1H-indole-2-carboxylic acid propan-2-yl ester Chemical compound C1=CC=C2NC(C(=O)OC(C)C)=CC2=C1OCC(O)CNC(C)(C)C SJYFDORQYYEJLB-UHFFFAOYSA-N 0.000 description 1
- OCXLBOACUPDBRL-UHFFFAOYSA-N 4-[4-(4-chlorobenzoyl)piperidin-1-yl]-1-(4-fluorophenyl)butan-1-one Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCC(C(=O)C=2C=CC(Cl)=CC=2)CC1 OCXLBOACUPDBRL-UHFFFAOYSA-N 0.000 description 1
- WPYGCZCMGMVGNO-UHFFFAOYSA-N 4-[4-[4-chloro-3-(trifluoromethyl)phenyl]-4-hydroxypiperidin-1-yl]-n,n-dimethyl-2,2-diphenylbutanamide Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C(=O)N(C)C)CCN(CC1)CCC1(O)C1=CC=C(Cl)C(C(F)(F)F)=C1 WPYGCZCMGMVGNO-UHFFFAOYSA-N 0.000 description 1
- DWUQCHRDSFIOAG-UHFFFAOYSA-N 4-[4-[bis(4-fluorophenyl)methyl]piperazin-1-yl]-1-(4-tert-butylphenyl)butan-1-ol Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCN(C(C=2C=CC(F)=CC=2)C=2C=CC(F)=CC=2)CC1 DWUQCHRDSFIOAG-UHFFFAOYSA-N 0.000 description 1
- YKGYIDJEEQRWQH-UHFFFAOYSA-N 4-[6-(diaminomethylideneamino)-1-oxohexoxy]benzoic acid ethyl ester Chemical compound CCOC(=O)C1=CC=C(OC(=O)CCCCCN=C(N)N)C=C1 YKGYIDJEEQRWQH-UHFFFAOYSA-N 0.000 description 1
- OYGDOCFZQVGFIP-UHFFFAOYSA-N 4-[8-fluoro-5-(4-fluorophenyl)-3,4-dihydro-1h-pyrido[4,3-b]indol-2-yl]-1-(4-fluorophenyl)butan-1-ol Chemical compound C=1C=C(F)C=CC=1C(O)CCCN(C1)CCC2=C1C1=CC(F)=CC=C1N2C1=CC=C(F)C=C1 OYGDOCFZQVGFIP-UHFFFAOYSA-N 0.000 description 1
- HLDUHCYBUVVDOT-GZBOUJLJSA-N 4-[[(3r,4s,5r)-3,4,5-trihydroxyoxan-2-yl]amino]benzoic acid Chemical compound O[C@@H]1[C@@H](O)[C@H](O)COC1NC1=CC=C(C(O)=O)C=C1 HLDUHCYBUVVDOT-GZBOUJLJSA-N 0.000 description 1
- XTWZHJXJIIUEJP-UHFFFAOYSA-N 4-acetamidobenzoic acid;2-(dimethylamino)ethanol Chemical compound CN(C)CCO.CC(=O)NC1=CC=C(C(O)=O)C=C1 XTWZHJXJIIUEJP-UHFFFAOYSA-N 0.000 description 1
- GIMSJJHKKXRFGV-BYPJNBLXSA-N 4-amino-1-[(2r,3s,4r,5r)-3-fluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-iodopyrimidin-2-one Chemical compound C1=C(I)C(N)=NC(=O)N1[C@H]1[C@@H](F)[C@H](O)[C@@H](CO)O1 GIMSJJHKKXRFGV-BYPJNBLXSA-N 0.000 description 1
- BTBFXLRQGPHRGT-UHFFFAOYSA-N 4-amino-2-butoxy-5-chloro-n-[1-(1,3-dioxolan-2-ylmethyl)piperidin-4-yl]benzamide Chemical compound CCCCOC1=CC(N)=C(Cl)C=C1C(=O)NC1CCN(CC2OCCO2)CC1 BTBFXLRQGPHRGT-UHFFFAOYSA-N 0.000 description 1
- WUIJEMRFIZSWPL-UHFFFAOYSA-N 4-amino-2-methoxy-5-nitro-n-[1-(oxolan-2-ylmethyl)piperidin-4-yl]benzamide Chemical compound COC1=CC(N)=C([N+]([O-])=O)C=C1C(=O)NC1CCN(CC2OCCC2)CC1 WUIJEMRFIZSWPL-UHFFFAOYSA-N 0.000 description 1
- GIYAQDDTCWHPPL-UHFFFAOYSA-N 4-amino-5-bromo-N-[2-(diethylamino)ethyl]-2-methoxybenzamide Chemical compound CCN(CC)CCNC(=O)C1=CC(Br)=C(N)C=C1OC GIYAQDDTCWHPPL-UHFFFAOYSA-N 0.000 description 1
- OVUIZYRVFCENSZ-UHFFFAOYSA-N 4-amino-5-bromo-n-[1-[(4-chlorophenyl)methyl]piperidin-4-yl]-2-methoxybenzamide Chemical compound COC1=CC(N)=C(Br)C=C1C(=O)NC1CCN(CC=2C=CC(Cl)=CC=2)CC1 OVUIZYRVFCENSZ-UHFFFAOYSA-N 0.000 description 1
- BVPWJMCABCPUQY-UHFFFAOYSA-N 4-amino-5-chloro-2-methoxy-N-[1-(phenylmethyl)-4-piperidinyl]benzamide Chemical compound COC1=CC(N)=C(Cl)C=C1C(=O)NC1CCN(CC=2C=CC=CC=2)CC1 BVPWJMCABCPUQY-UHFFFAOYSA-N 0.000 description 1
- QOVTVIYTBRHADL-UHFFFAOYSA-N 4-amino-6-(1,2,2-trichloroethenyl)benzene-1,3-disulfonamide Chemical compound NC1=CC(C(Cl)=C(Cl)Cl)=C(S(N)(=O)=O)C=C1S(N)(=O)=O QOVTVIYTBRHADL-UHFFFAOYSA-N 0.000 description 1
- VQWXCPNTBSKHJF-UHFFFAOYSA-N 4-amino-n,n-dimethyl-3-(4-methylpiperazine-1-carbonyl)benzenesulfonamide Chemical compound CN(C)S(=O)(=O)C1=CC=C(N)C(C(=O)N2CCN(C)CC2)=C1 VQWXCPNTBSKHJF-UHFFFAOYSA-N 0.000 description 1
- QGMGHALXLXKCBD-UHFFFAOYSA-N 4-amino-n-(2-aminophenyl)benzamide Chemical compound C1=CC(N)=CC=C1C(=O)NC1=CC=CC=C1N QGMGHALXLXKCBD-UHFFFAOYSA-N 0.000 description 1
- OPIGMRDAPFQALU-UHFFFAOYSA-N 4-amino-n-(5-propan-2-yl-1,3,4-thiadiazol-2-yl)benzenesulfonamide Chemical compound S1C(C(C)C)=NN=C1NS(=O)(=O)C1=CC=C(N)C=C1 OPIGMRDAPFQALU-UHFFFAOYSA-N 0.000 description 1
- HQFWVSGBVLEQGA-UHFFFAOYSA-N 4-aminobenzoic acid 3-(dibutylamino)propyl ester Chemical compound CCCCN(CCCC)CCCOC(=O)C1=CC=C(N)C=C1 HQFWVSGBVLEQGA-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- QGSGBBMKWJGCIE-UHFFFAOYSA-N 4-butyl-5-hydroxy-1,2-diphenylpyridazine-3,6-dione Chemical compound C=1C=CC=CC=1N1C(=O)C(CCCC)=C(O)C(=O)N1C1=CC=CC=C1 QGSGBBMKWJGCIE-UHFFFAOYSA-N 0.000 description 1
- JBVJKCHENYIOLW-UHFFFAOYSA-N 4-chloro-1,1,1,2,2,6-hexafluorohexane Chemical compound FCCC(Cl)CC(F)(F)C(F)(F)F JBVJKCHENYIOLW-UHFFFAOYSA-N 0.000 description 1
- OSDLLIBGSJNGJE-UHFFFAOYSA-N 4-chloro-3,5-dimethylphenol Chemical compound CC1=CC(O)=CC(C)=C1Cl OSDLLIBGSJNGJE-UHFFFAOYSA-N 0.000 description 1
- PNDYDXDNOWKEBO-UHFFFAOYSA-N 4-chloro-3-sulfamoyl-n-(2,4,6-trimethylpyridin-1-ium-1-yl)benzenecarboximidate Chemical compound CC1=CC(C)=CC(C)=[N+]1\N=C(/[O-])C1=CC=C(Cl)C(S(N)(=O)=O)=C1 PNDYDXDNOWKEBO-UHFFFAOYSA-N 0.000 description 1
- LBXHRAWDUMTPSE-AOOOYVTPSA-N 4-chloro-N-[(2S,6R)-2,6-dimethyl-1-piperidinyl]-3-sulfamoylbenzamide Chemical compound C[C@H]1CCC[C@@H](C)N1NC(=O)C1=CC=C(Cl)C(S(N)(=O)=O)=C1 LBXHRAWDUMTPSE-AOOOYVTPSA-N 0.000 description 1
- LKJLMYKYKMDEBY-UHFFFAOYSA-N 4-chloro-n-(2-hydroxyethyl)-n-[(2-methyl-3-bicyclo[2.2.1]heptanyl)methyl]benzamide Chemical compound CC1C(C2)CCC2C1CN(CCO)C(=O)C1=CC=C(Cl)C=C1 LKJLMYKYKMDEBY-UHFFFAOYSA-N 0.000 description 1
- REQFWARMBJWJAQ-UHFFFAOYSA-N 4-chloro-n-methyl-3-(methylsulfamoyl)benzamide Chemical compound CNC(=O)C1=CC=C(Cl)C(S(=O)(=O)NC)=C1 REQFWARMBJWJAQ-UHFFFAOYSA-N 0.000 description 1
- KKOWAYISKWGDBG-UHFFFAOYSA-N 4-deoxypyridoxine Chemical compound CC1=NC=C(CO)C(C)=C1O KKOWAYISKWGDBG-UHFFFAOYSA-N 0.000 description 1
- LQVMQEYROPXMQH-UHFFFAOYSA-N 4-dibenzofuran-2-yl-4-oxobutanoic acid Chemical compound C1=CC=C2C3=CC(C(=O)CCC(=O)O)=CC=C3OC2=C1 LQVMQEYROPXMQH-UHFFFAOYSA-N 0.000 description 1
- WYOMHOATUARGQV-UHFFFAOYSA-N 4-ethyl-3-methyl-3-phenylpyrrolidine-2,5-dione Chemical compound CCC1C(=O)NC(=O)C1(C)C1=CC=CC=C1 WYOMHOATUARGQV-UHFFFAOYSA-N 0.000 description 1
- ORRZGUBHBVWWOP-UHFFFAOYSA-N 4-ethyl-4-methylpiperidine-2,6-dione Chemical compound CCC1(C)CC(=O)NC(=O)C1 ORRZGUBHBVWWOP-UHFFFAOYSA-N 0.000 description 1
- SQHQDVNANXVULC-SNVBAGLBSA-N 4-hydroxy-3-[(1R)-1-hydroxypentyl]benzoic acid Chemical compound CCCC[C@@H](O)C1=CC(C(O)=O)=CC=C1O SQHQDVNANXVULC-SNVBAGLBSA-N 0.000 description 1
- SGOOQMRIPALTEL-UHFFFAOYSA-N 4-hydroxy-N,1-dimethyl-2-oxo-N-phenyl-3-quinolinecarboxamide Chemical compound OC=1C2=CC=CC=C2N(C)C(=O)C=1C(=O)N(C)C1=CC=CC=C1 SGOOQMRIPALTEL-UHFFFAOYSA-N 0.000 description 1
- LZCBNYVJTNCPDR-UHFFFAOYSA-N 4-methoxy-n-[5-(2-methylpropyl)-1,3,4-thiadiazol-2-yl]benzenesulfonamide Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)NC1=NN=C(CC(C)C)S1 LZCBNYVJTNCPDR-UHFFFAOYSA-N 0.000 description 1
- FLDROJIECSYZST-UHFFFAOYSA-N 4-methyl-2-[(4-methyl-3-nitropyridin-2-yl)disulfanyl]-3-nitropyridine Chemical compound CC1=CC=NC(SSC=2C(=C(C)C=CN=2)[N+]([O-])=O)=C1[N+]([O-])=O FLDROJIECSYZST-UHFFFAOYSA-N 0.000 description 1
- AUUADVNHIYKUBA-UHFFFAOYSA-N 4-methyl-n,n-dipropylbenzenesulfonamide Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C)C=C1 AUUADVNHIYKUBA-UHFFFAOYSA-N 0.000 description 1
- USABWIQJRBGMKQ-UHFFFAOYSA-N 4-n-(7-chloroquinolin-4-yl)-1-n,1-n-diethylpentane-1,4-diamine;8-hydroxy-7-iodoquinoline-5-sulfonic acid Chemical compound C1=CN=C2C(O)=C(I)C=C(S(O)(=O)=O)C2=C1.C1=CN=C2C(O)=C(I)C=C(S(O)(=O)=O)C2=C1.ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 USABWIQJRBGMKQ-UHFFFAOYSA-N 0.000 description 1
- RVMGXWBCQGAWBR-UHFFFAOYSA-N 4-oxo-1-benzopyran-2-carboxylic acid Chemical compound C1=CC=C2OC(C(=O)O)=CC(=O)C2=C1 RVMGXWBCQGAWBR-UHFFFAOYSA-N 0.000 description 1
- KWFFVBCJCQQAIV-UHFFFAOYSA-N 4-phenylcyclohexan-1-amine;2-(4-phenylphenyl)butanoic acid Chemical compound C1CC(N)CCC1C1=CC=CC=C1.C1=CC(C(C(O)=O)CC)=CC=C1C1=CC=CC=C1 KWFFVBCJCQQAIV-UHFFFAOYSA-N 0.000 description 1
- VQIAWQKBOAUGHE-UHFFFAOYSA-N 4-propylheptan-4-amine Chemical compound CCCC(N)(CCC)CCC VQIAWQKBOAUGHE-UHFFFAOYSA-N 0.000 description 1
- CXSJGNHRBWJXEA-UHFFFAOYSA-N 5,12-dihydrophthalazino[3,2-b]phthalazine-7,14-dione Chemical compound C1C2=CC=CC=C2C(=O)N2N1C(=O)C1=CC=CC=C1C2 CXSJGNHRBWJXEA-UHFFFAOYSA-N 0.000 description 1
- JYJFNDQBESEHJQ-UHFFFAOYSA-N 5,5-dimethyloxazolidine-2,4-dione Chemical compound CC1(C)OC(=O)NC1=O JYJFNDQBESEHJQ-UHFFFAOYSA-N 0.000 description 1
- FEJIIZAOQRTGPC-UHFFFAOYSA-N 5,5-diphenylimidazolidin-4-one Chemical compound O=C1NCNC1(C=1C=CC=CC=1)C1=CC=CC=C1 FEJIIZAOQRTGPC-UHFFFAOYSA-N 0.000 description 1
- ZEHAPDWJZZTSOT-UHFFFAOYSA-N 5,6-dimethoxy-4-methyl-3h-quinazolin-2-one;hydrochloride Chemical compound Cl.N1C(=O)N=C(C)C2=C(OC)C(OC)=CC=C21 ZEHAPDWJZZTSOT-UHFFFAOYSA-N 0.000 description 1
- GZSOSUNBTXMUFQ-NJGQXECBSA-N 5,7,3'-Trihydroxy-4'-methoxyflavone 7-O-rutinoside Natural products O(C[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@H](Oc2cc(O)c3C(=O)C=C(c4cc(O)c(OC)cc4)Oc3c2)O1)[C@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@H](C)O1 GZSOSUNBTXMUFQ-NJGQXECBSA-N 0.000 description 1
- PJJGZPJJTHBVMX-UHFFFAOYSA-N 5,7-Dihydroxyisoflavone Chemical compound C=1C(O)=CC(O)=C(C2=O)C=1OC=C2C1=CC=CC=C1 PJJGZPJJTHBVMX-UHFFFAOYSA-N 0.000 description 1
- BNACJQWJZKPAPV-UHFFFAOYSA-N 5,7-dibromo-2-methylquinolin-8-ol Chemical compound BrC1=CC(Br)=C(O)C2=NC(C)=CC=C21 BNACJQWJZKPAPV-UHFFFAOYSA-N 0.000 description 1
- ZDASUJMDVPTNTF-UHFFFAOYSA-N 5,7-dibromo-8-quinolinol Chemical compound C1=CN=C2C(O)=C(Br)C=C(Br)C2=C1 ZDASUJMDVPTNTF-UHFFFAOYSA-N 0.000 description 1
- YIMUZODUAWKOQL-UHFFFAOYSA-N 5-(1-benzylimidazol-2-yl)-2h-tetrazole Chemical compound C1=CN=C(C2=NNN=N2)N1CC1=CC=CC=C1 YIMUZODUAWKOQL-UHFFFAOYSA-N 0.000 description 1
- ALIOEEFZPKWPAV-UHFFFAOYSA-N 5-(1-methylpiperidin-4-ylidene)chromeno[2,3-b]pyridine Chemical compound C1CN(C)CCC1=C1C2=CC=CN=C2OC2=CC=CC=C21 ALIOEEFZPKWPAV-UHFFFAOYSA-N 0.000 description 1
- PXLPCZJACKUXGP-UHFFFAOYSA-N 5-(3,4-dichlorophenyl)-6-ethylpyrimidine-2,4-diamine Chemical compound CCC1=NC(N)=NC(N)=C1C1=CC=C(Cl)C(Cl)=C1 PXLPCZJACKUXGP-UHFFFAOYSA-N 0.000 description 1
- HMKQACOWZYBTIW-UHFFFAOYSA-N 5-(3-chloropropyl)-4-methyl-1,3-thiazole Chemical compound CC=1N=CSC=1CCCCl HMKQACOWZYBTIW-UHFFFAOYSA-N 0.000 description 1
- VHWFITPGPFLBGT-UHFFFAOYSA-N 5-(3-pyridinylmethyl)-2-benzofurancarboxylic acid Chemical compound C=1C=C2OC(C(=O)O)=CC2=CC=1CC1=CC=CN=C1 VHWFITPGPFLBGT-UHFFFAOYSA-N 0.000 description 1
- HEOZYYOUKGGSBJ-UHFFFAOYSA-N 5-(4-methoxybenzoyl)-2,3-dihydro-1h-pyrrolizine-1-carboxylic acid Chemical compound C1=CC(OC)=CC=C1C(=O)C1=CC=C2N1CCC2C(O)=O HEOZYYOUKGGSBJ-UHFFFAOYSA-N 0.000 description 1
- QNWOSJAGFSUDFE-UHFFFAOYSA-N 5-(aminooxymethyl)-2-bromophenol Chemical compound NOCC1=CC=C(Br)C(O)=C1 QNWOSJAGFSUDFE-UHFFFAOYSA-N 0.000 description 1
- YVQVOQKFMFRVGR-VGOFMYFVSA-N 5-(morpholin-4-ylmethyl)-3-[(e)-(5-nitrofuran-2-yl)methylideneamino]-1,3-oxazolidin-2-one Chemical compound O1C([N+](=O)[O-])=CC=C1\C=N\N1C(=O)OC(CN2CCOCC2)C1 YVQVOQKFMFRVGR-VGOFMYFVSA-N 0.000 description 1
- LSLYOANBFKQKPT-DIFFPNOSSA-N 5-[(1r)-1-hydroxy-2-[[(2r)-1-(4-hydroxyphenyl)propan-2-yl]amino]ethyl]benzene-1,3-diol Chemical compound C([C@@H](C)NC[C@H](O)C=1C=C(O)C=C(O)C=1)C1=CC=C(O)C=C1 LSLYOANBFKQKPT-DIFFPNOSSA-N 0.000 description 1
- NMARPFMJVCXSAV-UHFFFAOYSA-N 5-[(3,5-diethoxy-4-pyrrol-1-ylphenyl)methyl]pyrimidine-2,4-diamine Chemical compound C=1C(OCC)=C(N2C=CC=C2)C(OCC)=CC=1CC1=CN=C(N)N=C1N NMARPFMJVCXSAV-UHFFFAOYSA-N 0.000 description 1
- DNXDWMHPTVQBIP-ZQIUZPCESA-N 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]-n-[6-[(2-iodoacetyl)amino]hexyl]pentanamide Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)NCCCCCCNC(=O)CI)SC[C@@H]21 DNXDWMHPTVQBIP-ZQIUZPCESA-N 0.000 description 1
- QLPHBNRMJLFRGO-YDHSSHFGSA-N 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]-n-[6-[3-(pyridin-2-yldisulfanyl)propanoylamino]hexyl]pentanamide Chemical compound C([C@H]1[C@H]2NC(=O)N[C@H]2CS1)CCCC(=O)NCCCCCCNC(=O)CCSSC1=CC=CC=N1 QLPHBNRMJLFRGO-YDHSSHFGSA-N 0.000 description 1
- CIVGYTYIDWRBQU-UFLZEWODSA-N 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoic acid;pyrrole-2,5-dione Chemical group O=C1NC(=O)C=C1.N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 CIVGYTYIDWRBQU-UFLZEWODSA-N 0.000 description 1
- AUTKZDGLQYYXLZ-UHFFFAOYSA-N 5-[1-hydroxy-2-(3-phenylpropylamino)propyl]-2-methylphenol Chemical compound C=1C=C(C)C(O)=CC=1C(O)C(C)NCCCC1=CC=CC=C1 AUTKZDGLQYYXLZ-UHFFFAOYSA-N 0.000 description 1
- RRKXWTIXDRQLHE-UHFFFAOYSA-N 5-[2-(diethylamino)ethyl]-4-(4-fluorophenyl)-3h-1,3-oxazol-2-one Chemical compound O1C(=O)NC(C=2C=CC(F)=CC=2)=C1CCN(CC)CC RRKXWTIXDRQLHE-UHFFFAOYSA-N 0.000 description 1
- QPGGEKPRGVJKQB-UHFFFAOYSA-N 5-[2-(dimethylamino)ethyl]-11-methyl-6-benzo[b][1,4]benzodiazepinone Chemical compound O=C1N(CCN(C)C)C2=CC=CC=C2N(C)C2=CC=CC=C21 QPGGEKPRGVJKQB-UHFFFAOYSA-N 0.000 description 1
- ODACNRQBNVVGAI-UHFFFAOYSA-N 5-[2-chloroethyl(2-fluoroethyl)amino]-6-methyl-1h-pyrimidine-2,4-dione Chemical compound CC=1NC(=O)NC(=O)C=1N(CCF)CCCl ODACNRQBNVVGAI-UHFFFAOYSA-N 0.000 description 1
- TVPFRFLDGAWGAM-UHFFFAOYSA-N 5-[3-(dimethylamino)propyl]phenanthridin-6-one Chemical compound C1=CC=C2C(=O)N(CCCN(C)C)C3=CC=CC=C3C2=C1 TVPFRFLDGAWGAM-UHFFFAOYSA-N 0.000 description 1
- IXJJPGSXJXQFGI-UHFFFAOYSA-N 5-[3-[2-(3,4-dimethoxyphenyl)ethylamino]-2-hydroxypropoxy]-8-(2-oxopropoxy)-3,4-dihydro-1h-quinolin-2-one Chemical compound C1=C(OC)C(OC)=CC=C1CCNCC(O)COC1=CC=C(OCC(C)=O)C2=C1CCC(=O)N2 IXJJPGSXJXQFGI-UHFFFAOYSA-N 0.000 description 1
- DMDLRGRLIWYPQF-UHFFFAOYSA-N 5-[5-(3-phenylpropyl)thiophen-2-yl]pentanoic acid Chemical compound S1C(CCCCC(=O)O)=CC=C1CCCC1=CC=CC=C1 DMDLRGRLIWYPQF-UHFFFAOYSA-N 0.000 description 1
- BPEQAFGUCPPRIB-UHFFFAOYSA-M 5-[[5-(2-chloroethyl)-4-methyl-1,3-thiazol-3-ium-3-yl]methyl]-2-methylpyrimidin-4-amine;chloride Chemical compound [Cl-].CC1=C(CCCl)SC=[N+]1CC1=CN=C(C)N=C1N BPEQAFGUCPPRIB-UHFFFAOYSA-M 0.000 description 1
- SOQLNFHOYKNPDY-UHFFFAOYSA-N 5-acetyl-n-[(1-ethylpyrrolidin-2-yl)methyl]-2-methoxybenzamide Chemical compound CCN1CCCC1CNC(=O)C1=CC(C(C)=O)=CC=C1OC SOQLNFHOYKNPDY-UHFFFAOYSA-N 0.000 description 1
- GQCIBLGQUDFKGX-WLHGVMLRSA-M 5-amino-2-[(e)-2-(4-amino-2-sulfophenyl)ethenyl]benzenesulfonate;(2,4-dichlorophenoxy)methyl-dimethyl-octylazanium Chemical compound CCCCCCCC[N+](C)(C)COC1=CC=C(Cl)C=C1Cl.OS(=O)(=O)C1=CC(N)=CC=C1\C=C\C1=CC=C(N)C=C1S([O-])(=O)=O GQCIBLGQUDFKGX-WLHGVMLRSA-M 0.000 description 1
- UPALIKSFLSVKIS-UHFFFAOYSA-N 5-amino-2-[2-(dimethylamino)ethyl]benzo[de]isoquinoline-1,3-dione Chemical compound NC1=CC(C(N(CCN(C)C)C2=O)=O)=C3C2=CC=CC3=C1 UPALIKSFLSVKIS-UHFFFAOYSA-N 0.000 description 1
- KEYYFWXKFRRLOZ-UHFFFAOYSA-N 5-bromo-2-hydroxy-3-methylbenzamide Chemical compound CC1=CC(Br)=CC(C(N)=O)=C1O KEYYFWXKFRRLOZ-UHFFFAOYSA-N 0.000 description 1
- CTFFKFYWSOSIAA-UHFFFAOYSA-N 5-bromo-n-(4-bromophenyl)-2-hydroxybenzamide Chemical compound OC1=CC=C(Br)C=C1C(=O)NC1=CC=C(Br)C=C1 CTFFKFYWSOSIAA-UHFFFAOYSA-N 0.000 description 1
- WOVKYSAHUYNSMH-RRKCRQDMSA-N 5-bromodeoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-RRKCRQDMSA-N 0.000 description 1
- DVEQCIBLXRSYPH-UHFFFAOYSA-N 5-butyl-1-cyclohexylbarbituric acid Chemical compound O=C1C(CCCC)C(=O)NC(=O)N1C1CCCCC1 DVEQCIBLXRSYPH-UHFFFAOYSA-N 0.000 description 1
- VWXGRWMELBPMCU-UHFFFAOYSA-N 5-chloro-1-[3-(dimethylamino)propyl]-3-phenylbenzimidazol-2-one Chemical compound O=C1N(CCCN(C)C)C2=CC=C(Cl)C=C2N1C1=CC=CC=C1 VWXGRWMELBPMCU-UHFFFAOYSA-N 0.000 description 1
- RXOBKRPNVDTZQF-UHFFFAOYSA-N 5-chloro-2-methoxy-n-[2-[4-(methylcarbamoylsulfamoyl)phenyl]ethyl]benzamide Chemical compound C1=CC(S(=O)(=O)NC(=O)NC)=CC=C1CCNC(=O)C1=CC(Cl)=CC=C1OC RXOBKRPNVDTZQF-UHFFFAOYSA-N 0.000 description 1
- IRAICTQDUFYXMD-UHFFFAOYSA-N 5-chloro-2-methoxy-n-[3-[4-(3-methylphenyl)piperazin-1-yl]propylcarbamoyl]benzamide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NC(=O)NCCCN1CCN(C=2C=C(C)C=CC=2)CC1 IRAICTQDUFYXMD-UHFFFAOYSA-N 0.000 description 1
- AADCDMQTJNYOSS-LBPRGKRZSA-N 5-chloro-3-ethyl-N-[[(2S)-1-ethyl-2-pyrrolidinyl]methyl]-2-hydroxy-6-methoxybenzamide Chemical compound CCN1CCC[C@H]1CNC(=O)C1=C(O)C(CC)=CC(Cl)=C1OC AADCDMQTJNYOSS-LBPRGKRZSA-N 0.000 description 1
- VZNJZQAFQDEJOO-UHFFFAOYSA-N 5-chloro-4-[[2-(4-chlorophenoxy)acetyl]amino]-n-[2-(diethylamino)ethyl]-2-methoxybenzamide Chemical compound C1=C(OC)C(C(=O)NCCN(CC)CC)=CC(Cl)=C1NC(=O)COC1=CC=C(Cl)C=C1 VZNJZQAFQDEJOO-UHFFFAOYSA-N 0.000 description 1
- FFPGKSCPXZYKFO-UHFFFAOYSA-N 5-chloro-6-cyclohexyl-3h-1-benzofuran-2-one Chemical compound ClC1=CC=2CC(=O)OC=2C=C1C1CCCCC1 FFPGKSCPXZYKFO-UHFFFAOYSA-N 0.000 description 1
- XFLJNCDBYCPPTE-UHFFFAOYSA-N 5-diazonio-6-oxo-1h-pyrimidin-2-olate Chemical compound [O-]C1=NC=C([N+]#N)C(=O)N1 XFLJNCDBYCPPTE-UHFFFAOYSA-N 0.000 description 1
- BITVSRNAFFZUFW-UHFFFAOYSA-N 5-ethyl-6-phenylphenanthridin-5-ium-3,8-diamine;chloride Chemical compound [Cl-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 BITVSRNAFFZUFW-UHFFFAOYSA-N 0.000 description 1
- IIRVYWCKYUQJCL-UHFFFAOYSA-N 5-ethyl-8-oxo-2,3-dihydrofuro[2,3-g]quinoline-7-carboxylic acid Chemical compound C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC2=C1CCO2 IIRVYWCKYUQJCL-UHFFFAOYSA-N 0.000 description 1
- TUTQORSNAITDSX-ONNFQVAWSA-N 5-methyl-3-[(e)-(5-nitrofuran-2-yl)methylideneamino]-1,3-oxazolidin-2-one Chemical compound O=C1OC(C)CN1\N=C\C1=CC=C([N+]([O-])=O)O1 TUTQORSNAITDSX-ONNFQVAWSA-N 0.000 description 1
- RZTAMFZIAATZDJ-HNNXBMFYSA-N 5-o-ethyl 3-o-methyl (4s)-4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OC)[C@@H]1C1=CC=CC(Cl)=C1Cl RZTAMFZIAATZDJ-HNNXBMFYSA-N 0.000 description 1
- AGNRWZQXNQBVQZ-UHFFFAOYSA-N 5-phenyl-3-(trichloromethylsulfanyl)-1,3,4-oxadiazol-2-one Chemical compound O1C(=O)N(SC(Cl)(Cl)Cl)N=C1C1=CC=CC=C1 AGNRWZQXNQBVQZ-UHFFFAOYSA-N 0.000 description 1
- KTPVSQNBBGUGAJ-UHFFFAOYSA-N 5-phenylpyrimido[4,5-d]pyrimidine-2,4,7-triamine Chemical compound N=1C(N)=NC2=NC(N)=NC(N)=C2C=1C1=CC=CC=C1 KTPVSQNBBGUGAJ-UHFFFAOYSA-N 0.000 description 1
- TZBDXWBBMOEVPI-XBQQDWOSSA-N 58524-83-7 Chemical compound O=C([C@]12[C@@]3(C)C[C@H](O)[C@]4(F)[C@@]5(C)C=CC(=O)C=C5[C@@H](F)C[C@H]4[C@@H]3C[C@H]1OC(O2)(C)C)COC(=O)C1CC1 TZBDXWBBMOEVPI-XBQQDWOSSA-N 0.000 description 1
- CBMYJHIOYJEBSB-YSZCXEEOSA-N 5alpha-androstane-3beta,17beta-diol Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 CBMYJHIOYJEBSB-YSZCXEEOSA-N 0.000 description 1
- LJVZNDLLRXZPNO-UHFFFAOYSA-N 6-(diethylaminomethyl)-3-methyl-2-phenylchromen-4-one Chemical compound CC=1C(=O)C2=CC(CN(CC)CC)=CC=C2OC=1C1=CC=CC=C1 LJVZNDLLRXZPNO-UHFFFAOYSA-N 0.000 description 1
- HCNBCFYKPSFHLH-UHFFFAOYSA-N 6-(furan-2-yl)pteridine-2,4,7-triamine Chemical compound NC1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CO1 HCNBCFYKPSFHLH-UHFFFAOYSA-N 0.000 description 1
- BFPYUXIFGJJYHU-AYSLTRBKSA-N 6-[(e)-1-phenylprop-1-enyl]-1-propan-2-ylsulfonylbenzimidazol-2-amine Chemical compound C=1C=C2N=C(N)N(S(=O)(=O)C(C)C)C2=CC=1C(=C/C)/C1=CC=CC=C1 BFPYUXIFGJJYHU-AYSLTRBKSA-N 0.000 description 1
- WGMYEOIMVYADRJ-UHFFFAOYSA-N 6-[2-(diethylamino)ethoxy]-N,N-dimethyl-1,3-benzothiazol-2-amine Chemical compound CCN(CC)CCOC1=CC=C2N=C(N(C)C)SC2=C1 WGMYEOIMVYADRJ-UHFFFAOYSA-N 0.000 description 1
- ZEWJVHFRQFCLFW-UHFFFAOYSA-N 6-[2-(dimethylamino)ethyl]pyrido[2,3-b][1,5]benzothiazepin-5-one Chemical compound O=C1N(CCN(C)C)C2=CC=CC=C2SC2=NC=CC=C21 ZEWJVHFRQFCLFW-UHFFFAOYSA-N 0.000 description 1
- KXBCLNRMQPRVTP-UHFFFAOYSA-N 6-amino-1,5-dihydroimidazo[4,5-c]pyridin-4-one Chemical compound O=C1NC(N)=CC2=C1N=CN2 KXBCLNRMQPRVTP-UHFFFAOYSA-N 0.000 description 1
- WYXSYVWAUAUWLD-SHUUEZRQSA-N 6-azauridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=N1 WYXSYVWAUAUWLD-SHUUEZRQSA-N 0.000 description 1
- JRXGULDSFFLUAO-UHFFFAOYSA-N 6-bromo-4,4-dimethyl-1h-3,1-benzoxazin-2-one Chemical compound C1=C(Br)C=C2C(C)(C)OC(=O)NC2=C1 JRXGULDSFFLUAO-UHFFFAOYSA-N 0.000 description 1
- KVAWWXSLBDVXHJ-UHFFFAOYSA-N 6-bromo-5-chloro-3h-1,3-benzoxazol-2-one Chemical compound C1=C(Br)C(Cl)=CC2=C1OC(=O)N2 KVAWWXSLBDVXHJ-UHFFFAOYSA-N 0.000 description 1
- BQVIONBNDNFCCP-UHFFFAOYSA-N 6-butyl-4,10-dioxo-1,7-dihydro-1,7-phenanthroline-2,8-dicarboxylic acid Chemical compound N1C(C(O)=O)=CC(=O)C2=C1C(CCCC)=CC1=C2NC(C(O)=O)=CC1=O BQVIONBNDNFCCP-UHFFFAOYSA-N 0.000 description 1
- BKYKPTRYDKTTJY-UHFFFAOYSA-N 6-chloro-3-(cyclopentylmethyl)-1,1-dioxo-3,4-dihydro-2H-1$l^{6},2,4-benzothiadiazine-7-sulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2CC1CCCC1 BKYKPTRYDKTTJY-UHFFFAOYSA-N 0.000 description 1
- WFJWTVCUKPPEOR-UHFFFAOYSA-N 6-chloro-3-[1-(2,3-dihydro-1,4-benzodioxin-3-ylmethyl)piperidin-4-yl]-1h-benzimidazol-2-one Chemical compound C1OC2=CC=CC=C2OC1CN(CC1)CCC1N1C2=CC=C(Cl)C=C2NC1=O WFJWTVCUKPPEOR-UHFFFAOYSA-N 0.000 description 1
- YIAHFLWLLVEPPS-UHFFFAOYSA-N 6-chloro-4-(2,3-dihydroxypropyl)-2-methyl-1,4-benzoxazin-3-one Chemical compound C1=C(Cl)C=C2N(CC(O)CO)C(=O)C(C)OC2=C1 YIAHFLWLLVEPPS-UHFFFAOYSA-N 0.000 description 1
- OAIZNWQBWDHNIH-UHFFFAOYSA-N 6-chloro-4-phenyl-1-(2,2,2-trifluoroethyl)quinazolin-2-one Chemical compound N=1C(=O)N(CC(F)(F)F)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 OAIZNWQBWDHNIH-UHFFFAOYSA-N 0.000 description 1
- ZGOWZGZRTGGMMM-UHFFFAOYSA-N 6-chloro-n-ethyl-4-phenyl-4h-3,1-benzothiazin-2-amine Chemical compound S1C(NCC)=NC2=CC=C(Cl)C=C2C1C1=CC=CC=C1 ZGOWZGZRTGGMMM-UHFFFAOYSA-N 0.000 description 1
- ZNXAJGZPUQOEDZ-UHFFFAOYSA-N 6-ethyl-4,5,7,8-tetrahydro-[1,3]oxazolo[4,5-d]azepin-2-amine Chemical compound C1CN(CC)CCC2=C1OC(N)=N2 ZNXAJGZPUQOEDZ-UHFFFAOYSA-N 0.000 description 1
- UPMCDOMOBNMTPH-UHFFFAOYSA-N 6-phenyl-5,6-dihydroimidazo[2,1-b][1,3]thiazole Chemical compound N1=C2SC=CN2CC1C1=CC=CC=C1 UPMCDOMOBNMTPH-UHFFFAOYSA-N 0.000 description 1
- RFOLEEQBNNSICL-UHFFFAOYSA-N 6-thiophen-2-yl-5,6-dihydroimidazo[2,1-b][1,3]thiazole Chemical compound N1=C2SC=CN2CC1C1=CC=CS1 RFOLEEQBNNSICL-UHFFFAOYSA-N 0.000 description 1
- IUIYEHXOIMMQJY-NGXOUOCZSA-N 60135-22-0 Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)C(OC)OC)[C@@]2(C)C[C@@H]1O IUIYEHXOIMMQJY-NGXOUOCZSA-N 0.000 description 1
- MYYIMZRZXIQBGI-HVIRSNARSA-N 6alpha-Fluoroprednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3C[C@H](F)C2=C1 MYYIMZRZXIQBGI-HVIRSNARSA-N 0.000 description 1
- UBGCCYGMLOBJOJ-UHFFFAOYSA-N 7-(1,4-diazabicyclo[3.2.2]nonan-4-yl)-1-ethyl-6-fluoro-4-oxoquinoline-3-carboxylic acid Chemical compound C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCN2CCC1CC2 UBGCCYGMLOBJOJ-UHFFFAOYSA-N 0.000 description 1
- NWPRCRWQMGIBOT-UHFFFAOYSA-N 7-(2-hydroxyethyl)-1,3-dimethylpurine-2,6-dione Chemical compound O=C1N(C)C(=O)N(C)C2=C1N(CCO)C=N2 NWPRCRWQMGIBOT-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- OQMZNAMGEHIHNN-UHFFFAOYSA-N 7-Dehydrostigmasterol Natural products C1C(O)CCC2(C)C(CCC3(C(C(C)C=CC(CC)C(C)C)CCC33)C)C3=CC=C21 OQMZNAMGEHIHNN-UHFFFAOYSA-N 0.000 description 1
- KTEHTTNELUAQFC-AHRHGKHWSA-N 7-[(1r,2r)-2-[(e,3s)-3-hydroxy-3-methyloct-1-enyl]-5-oxocyclopentyl]heptanoic acid Chemical compound CCCCC[C@](C)(O)\C=C\[C@H]1CCC(=O)[C@@H]1CCCCCCC(O)=O KTEHTTNELUAQFC-AHRHGKHWSA-N 0.000 description 1
- JERCJPRNXXOPNI-FYQIFJIOSA-N 7-[(1r,2s)-2-(3-hydroxy-3-methyloctyl)-5-oxocyclopentyl]heptanoic acid Chemical compound CCCCCC(C)(O)CC[C@H]1CCC(=O)[C@@H]1CCCCCCC(O)=O JERCJPRNXXOPNI-FYQIFJIOSA-N 0.000 description 1
- PMEYQPKJAPXGPS-UHFFFAOYSA-N 7-[2-[4-(4-fluorobenzoyl)piperidin-1-yl]ethyl]-1,3-dimethylpurine-2,6-dione Chemical compound C1=2C(=O)N(C)C(=O)N(C)C=2N=CN1CCN(CC1)CCC1C(=O)C1=CC=C(F)C=C1 PMEYQPKJAPXGPS-UHFFFAOYSA-N 0.000 description 1
- IPSYNNODBHPDFB-UHFFFAOYSA-N 7-[3-[4-(4-fluorobenzoyl)piperidin-1-yl]propyl]-1,3-dimethylpurine-2,6-dione Chemical compound C1=2C(=O)N(C)C(=O)N(C)C=2N=CN1CCCN(CC1)CCC1C(=O)C1=CC=C(F)C=C1 IPSYNNODBHPDFB-UHFFFAOYSA-N 0.000 description 1
- VUCZRCBQVXTIIV-UHFFFAOYSA-N 7-[[4-(dimethylamino)phenyl]methyl]-1,3-dimethylpurine-2,6-dione Chemical compound C1=CC(N(C)C)=CC=C1CN1C(C(=O)N(C)C(=O)N2C)=C2N=C1 VUCZRCBQVXTIIV-UHFFFAOYSA-N 0.000 description 1
- ICLLENNEPRZLAP-UHFFFAOYSA-N 7-bromo-3,4-dihydro-1h-isoquinoline-2-carboximidamide Chemical compound C1=C(Br)C=C2CN(C(=N)N)CCC2=C1 ICLLENNEPRZLAP-UHFFFAOYSA-N 0.000 description 1
- AWHZKKVSUJJVNL-UHFFFAOYSA-N 7-chloro-10-hydroxy-3-[4-(trifluoromethyl)phenyl]-3,4-dihydro-2h-acridine-1,9-dione Chemical compound C1C(=O)C=2C(=O)C3=CC(Cl)=CC=C3N(O)C=2CC1C1=CC=C(C(F)(F)F)C=C1 AWHZKKVSUJJVNL-UHFFFAOYSA-N 0.000 description 1
- ATAJVFBUUIBIEO-UHFFFAOYSA-N 7-chloro-2,3-dihydro-1h-inden-4-ol Chemical compound OC1=CC=C(Cl)C2=C1CCC2 ATAJVFBUUIBIEO-UHFFFAOYSA-N 0.000 description 1
- UIEGCHMMGBNMIL-UHFFFAOYSA-N 7-chloro-4-hydroxy-2,3-dihydro-1h-indene-5-carboxylic acid Chemical compound OC(=O)C1=CC(Cl)=C2CCCC2=C1O UIEGCHMMGBNMIL-UHFFFAOYSA-N 0.000 description 1
- ANOGOQXCGBMIJV-UHFFFAOYSA-N 7-chloro-n-(3,4-dichlorophenyl)-5-hydroxy-1,1-dioxo-2,3-dihydro-1$l^{6}-benzothiepine-4-carboxamide Chemical compound C1CS(=O)(=O)C2=CC=C(Cl)C=C2C(O)=C1C(=O)NC1=CC=C(Cl)C(Cl)=C1 ANOGOQXCGBMIJV-UHFFFAOYSA-N 0.000 description 1
- CDMRMPWJHYFXJJ-UHFFFAOYSA-N 7-methylpyrazolo[1,5-a][1,3,5]triazine-2,4-diamine Chemical compound N1=C(N)N=C(N)N2N=C(C)C=C21 CDMRMPWJHYFXJJ-UHFFFAOYSA-N 0.000 description 1
- OZSPQIXKOVJJGE-UHFFFAOYSA-N 8-(2-ethoxyethyl)-7-phenyl-[1,2,4]triazolo[1,5-c]pyrimidin-5-amine Chemical compound N1=C(N)N2N=CN=C2C(CCOCC)=C1C1=CC=CC=C1 OZSPQIXKOVJJGE-UHFFFAOYSA-N 0.000 description 1
- FVNFBBAOMBJTST-UHFFFAOYSA-N 8-(2-phenylethyl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one Chemical compound O1C(=O)NCC11CCN(CCC=2C=CC=CC=2)CC1 FVNFBBAOMBJTST-UHFFFAOYSA-N 0.000 description 1
- WXIGSVFQTLVMQM-UHFFFAOYSA-N 8-(trifluoromethyl)-10h-phenothiazine-1-carboxylic acid Chemical compound S1C2=CC=C(C(F)(F)F)C=C2NC2=C1C=CC=C2C(=O)O WXIGSVFQTLVMQM-UHFFFAOYSA-N 0.000 description 1
- QOYHHIBFXOOADH-UHFFFAOYSA-N 8-[4,4-bis(4-fluorophenyl)butyl]-1-phenyl-1,3,8-triazaspiro[4.5]decan-4-one Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)CCCN1CCC2(C(NCN2C=2C=CC=CC=2)=O)CC1 QOYHHIBFXOOADH-UHFFFAOYSA-N 0.000 description 1
- VQSXXDXNONIOGW-UHFFFAOYSA-N 8-[[bis(2-hydroxyethyl)amino]methyl]-6,7-dihydroxy-4-methylchromen-2-one Chemical compound OCCN(CCO)CC1=C(O)C(O)=CC2=C1OC(=O)C=C2C VQSXXDXNONIOGW-UHFFFAOYSA-N 0.000 description 1
- WCTGYFWVYBPRGF-UHFFFAOYSA-N 8-chloro-2-methyl-5-[2-(6-methylpyridin-3-yl)ethyl]-3,4-dihydro-1h-pyrido[4,3-b]indole Chemical compound C1N(C)CCC2=C1C1=CC(Cl)=CC=C1N2CCC1=CC=C(C)N=C1 WCTGYFWVYBPRGF-UHFFFAOYSA-N 0.000 description 1
- RZAGBWYFAWQKAE-UHFFFAOYSA-N 8-chloro-6-[(1-propan-2-yl-4,5-dihydroimidazol-2-yl)methyl]-2,3,4,5-tetrahydro-1,6-benzothiazocine Chemical compound CC(C)N1CCN=C1CN1C2=CC(Cl)=CC=C2SCCCC1 RZAGBWYFAWQKAE-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- RRTGJSZJWLUVRE-UHFFFAOYSA-N 9-[3-(tert-butylamino)-2-hydroxypropoxy]-4-hydroxy-7-methylfuro[3,2-g]chromen-5-one Chemical compound CC(C)(C)NCC(O)COC1=C2OC(C)=CC(=O)C2=C(O)C2=C1OC=C2 RRTGJSZJWLUVRE-UHFFFAOYSA-N 0.000 description 1
- APDNCRBSYUOIKC-UHFFFAOYSA-N 9-ethyl-3h-purine-6-thione Chemical compound N1C=NC(=S)C2=C1N(CC)C=N2 APDNCRBSYUOIKC-UHFFFAOYSA-N 0.000 description 1
- DPSPPJIUMHPXMA-UHFFFAOYSA-N 9-fluoro-5-methyl-1-oxo-6,7-dihydro-1H,5H-pyrido[3,2,1-ij]quinoline-2-carboxylic acid Chemical compound C1CC(C)N2C=C(C(O)=O)C(=O)C3=C2C1=CC(F)=C3 DPSPPJIUMHPXMA-UHFFFAOYSA-N 0.000 description 1
- WRDABNWSWOHGMS-UHFFFAOYSA-N AEBSF hydrochloride Chemical compound Cl.NCCC1=CC=C(S(F)(=O)=O)C=C1 WRDABNWSWOHGMS-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- UCTWMZQNUQWSLP-UHFFFAOYSA-N Adrenaline Natural products CNCC(O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-UHFFFAOYSA-N 0.000 description 1
- 102100036601 Aggrecan core protein Human genes 0.000 description 1
- 108010067219 Aggrecans Proteins 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- KHOITXIGCFIULA-UHFFFAOYSA-N Alophen Chemical compound C1=CC(OC(=O)C)=CC=C1C(C=1N=CC=CC=1)C1=CC=C(OC(C)=O)C=C1 KHOITXIGCFIULA-UHFFFAOYSA-N 0.000 description 1
- 229920001450 Alpha-Cyclodextrin Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- RLFWWDJHLFCNIJ-UHFFFAOYSA-N Aminoantipyrine Natural products CN1C(C)=C(N)C(=O)N1C1=CC=CC=C1 RLFWWDJHLFCNIJ-UHFFFAOYSA-N 0.000 description 1
- 102000004400 Aminopeptidases Human genes 0.000 description 1
- 108090000915 Aminopeptidases Proteins 0.000 description 1
- OVCDSSHSILBFBN-UHFFFAOYSA-N Amodiaquine Chemical compound C1=C(O)C(CN(CC)CC)=CC(NC=2C3=CC=C(Cl)C=C3N=CC=2)=C1 OVCDSSHSILBFBN-UHFFFAOYSA-N 0.000 description 1
- CAVZBWFUMSXZFB-LJWNLINESA-N Amogastrin Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)OC(C)(C)CC)C(N)=O)C1=CC=CC=C1 CAVZBWFUMSXZFB-LJWNLINESA-N 0.000 description 1
- 102000013455 Amyloid beta-Peptides Human genes 0.000 description 1
- 108010090849 Amyloid beta-Peptides Proteins 0.000 description 1
- 229920000945 Amylopectin Polymers 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 102100022987 Angiogenin Human genes 0.000 description 1
- 102000009840 Angiopoietins Human genes 0.000 description 1
- 108010009906 Angiopoietins Proteins 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- WZPBZJONDBGPKJ-UHFFFAOYSA-N Antibiotic SQ 26917 Natural products O=C1N(S(O)(=O)=O)C(C)C1NC(=O)C(=NOC(C)(C)C(O)=O)C1=CSC(N)=N1 WZPBZJONDBGPKJ-UHFFFAOYSA-N 0.000 description 1
- 102100040199 Apolipoprotein B receptor Human genes 0.000 description 1
- 102100029470 Apolipoprotein E Human genes 0.000 description 1
- 101710095339 Apolipoprotein E Proteins 0.000 description 1
- 102000007592 Apolipoproteins Human genes 0.000 description 1
- 108010071619 Apolipoproteins Proteins 0.000 description 1
- OZDNDGXASTWERN-CTNGQTDRSA-N Apovincamine Chemical compound C1=CC=C2C(CCN3CCC4)=C5[C@@H]3[C@]4(CC)C=C(C(=O)OC)N5C2=C1 OZDNDGXASTWERN-CTNGQTDRSA-N 0.000 description 1
- QTGIAADRBBLJGA-UHFFFAOYSA-N Articaine Chemical compound CCCNC(C)C(=O)NC=1C(C)=CSC=1C(=O)OC QTGIAADRBBLJGA-UHFFFAOYSA-N 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- BHELIUBJHYAEDK-OAIUPTLZSA-N Aspoxicillin Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3[C@H](C(C)(C)S[C@@H]32)C(O)=O)=O)NC(=O)[C@H](N)CC(=O)NC)=CC=C(O)C=C1 BHELIUBJHYAEDK-OAIUPTLZSA-N 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- YXSLJKQTIDHPOT-UHFFFAOYSA-N Atracurium Dibesylate Chemical compound C1=C(OC)C(OC)=CC=C1CC1[N+](CCC(=O)OCCCCCOC(=O)CC[N+]2(C)C(C3=CC(OC)=C(OC)C=C3CC2)CC=2C=C(OC)C(OC)=CC=2)(C)CCC2=CC(OC)=C(OC)C=C21 YXSLJKQTIDHPOT-UHFFFAOYSA-N 0.000 description 1
- 108010006835 Atrial Natriuretic Factor Receptors Proteins 0.000 description 1
- 229930003347 Atropine Natural products 0.000 description 1
- XIRGHRXBGGPPKY-UHFFFAOYSA-N Avilamycin-A Natural products COCC1OC(OC2C(C3OC4(OC3CO2)C2OCOC2C(O)(C(C)O4)C(C)=O)OC(=O)C(C)C)C(OC)C(O)C1OC(C1O)OC(C)C(OC)C1OC(OC(C)C1O2)CC1(C)OC2(OC1C)CC(O)C1OC(OC1C)CC(O)C1OC(=O)C1=C(C)C(Cl)=C(O)C(Cl)=C1OC XIRGHRXBGGPPKY-UHFFFAOYSA-N 0.000 description 1
- RQBNXPJPWKUTOG-UHFFFAOYSA-N Azabon Chemical compound C1=CC(N)=CC=C1S(=O)(=O)N1CC(CC2)CCC2C1 RQBNXPJPWKUTOG-UHFFFAOYSA-N 0.000 description 1
- MBUVEWMHONZEQD-UHFFFAOYSA-N Azeptin Chemical compound C1CN(C)CCCC1N1C(=O)C2=CC=CC=C2C(CC=2C=CC(Cl)=CC=2)=N1 MBUVEWMHONZEQD-UHFFFAOYSA-N 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 1
- BXTVQNYQYUTQAZ-UHFFFAOYSA-N BNPS-skatole Chemical compound N=1C2=CC=CC=C2C(C)(Br)C=1SC1=CC=CC=C1[N+]([O-])=O BXTVQNYQYUTQAZ-UHFFFAOYSA-N 0.000 description 1
- WOVKYSAHUYNSMH-UHFFFAOYSA-N BROMODEOXYURIDINE Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-UHFFFAOYSA-N 0.000 description 1
- KPYSYYIEGFHWSV-UHFFFAOYSA-N Baclofen Chemical compound OC(=O)CC(CN)C1=CC=C(Cl)C=C1 KPYSYYIEGFHWSV-UHFFFAOYSA-N 0.000 description 1
- SPFYMRJSYKOXGV-UHFFFAOYSA-N Baytril Chemical compound C1CN(CC)CCN1C(C(=C1)F)=CC2=C1C(=O)C(C(O)=O)=CN2C1CC1 SPFYMRJSYKOXGV-UHFFFAOYSA-N 0.000 description 1
- PYPJRLVCFAVWFR-UHFFFAOYSA-N Benapryzine Chemical compound C=1C=CC=CC=1C(O)(C(=O)OCCN(CC)CCC)C1=CC=CC=C1 PYPJRLVCFAVWFR-UHFFFAOYSA-N 0.000 description 1
- XPCFTKFZXHTYIP-PMACEKPBSA-N Benazepril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XPCFTKFZXHTYIP-PMACEKPBSA-N 0.000 description 1
- LQQIVYSCPWCSSD-UHFFFAOYSA-N Benzetimide Chemical compound O=C1NC(=O)CCC1(C=1C=CC=CC=1)C1CCN(CC=2C=CC=CC=2)CC1 LQQIVYSCPWCSSD-UHFFFAOYSA-N 0.000 description 1
- UYIFTLBWAOGQBI-BZDYCCQFSA-N Benzhormovarine Chemical compound C([C@@H]1[C@@H](C2=CC=3)CC[C@]4([C@H]1CC[C@@H]4O)C)CC2=CC=3OC(=O)C1=CC=CC=C1 UYIFTLBWAOGQBI-BZDYCCQFSA-N 0.000 description 1
- CULUWZNBISUWAS-UHFFFAOYSA-N Benznidazole Chemical compound [O-][N+](=O)C1=NC=CN1CC(=O)NCC1=CC=CC=C1 CULUWZNBISUWAS-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- VGGGPCQERPFHOB-UHFFFAOYSA-N Bestatin Natural products CC(C)CC(C(O)=O)NC(=O)C(O)C(N)CC1=CC=CC=C1 VGGGPCQERPFHOB-UHFFFAOYSA-N 0.000 description 1
- 102100030802 Beta-2-glycoprotein 1 Human genes 0.000 description 1
- 102000004954 Biglycan Human genes 0.000 description 1
- 108090001138 Biglycan Proteins 0.000 description 1
- QVZCXCJXTMIDME-UHFFFAOYSA-N Biopropazepan Trimethoxybenzoate Chemical compound COC1=C(OC)C(OC)=CC(C(=O)OCCCN2CCN(CCCOC(=O)C=3C=C(OC)C(OC)=C(OC)C=3)CCC2)=C1 QVZCXCJXTMIDME-UHFFFAOYSA-N 0.000 description 1
- IWXAZSAGYJHXPX-BCEWYCLDSA-N Bisbentiamine Chemical compound C=1C=CC=CC=1C(=O)OCC/C(SS\C(CCOC(=O)C=1C=CC=CC=1)=C(/C)N(CC=1C(=NC(C)=NC=1)N)C=O)=C(/C)N(C=O)CC1=CN=C(C)N=C1N IWXAZSAGYJHXPX-BCEWYCLDSA-N 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- IVFYLRMMHVYGJH-VLOLGRDOSA-N Bolasterone Chemical compound C1C[C@]2(C)[C@](O)(C)CC[C@H]2[C@@H]2[C@H](C)CC3=CC(=O)CC[C@]3(C)[C@H]21 IVFYLRMMHVYGJH-VLOLGRDOSA-N 0.000 description 1
- 102000001893 Bone Morphogenetic Protein Receptors Human genes 0.000 description 1
- 108010040422 Bone Morphogenetic Protein Receptors Proteins 0.000 description 1
- 239000004135 Bone phosphate Substances 0.000 description 1
- 101800004538 Bradykinin Proteins 0.000 description 1
- 102400000967 Bradykinin Human genes 0.000 description 1
- 244000056139 Brassica cretica Species 0.000 description 1
- 235000003351 Brassica cretica Nutrition 0.000 description 1
- 235000003343 Brassica rupestris Nutrition 0.000 description 1
- VMIYHDSEFNYJSL-UHFFFAOYSA-N Bromazepam Chemical compound C12=CC(Br)=CC=C2NC(=O)CN=C1C1=CC=CC=N1 VMIYHDSEFNYJSL-UHFFFAOYSA-N 0.000 description 1
- MMYHZEDBTMXYAF-UHFFFAOYSA-N Bromofenofos Chemical compound OC1=C(Br)C=C(Br)C=C1C1=CC(Br)=CC(Br)=C1OP(O)(O)=O MMYHZEDBTMXYAF-UHFFFAOYSA-N 0.000 description 1
- RKLNONIVDFXQRX-UHFFFAOYSA-N Bromperidol Chemical compound C1CC(O)(C=2C=CC(Br)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 RKLNONIVDFXQRX-UHFFFAOYSA-N 0.000 description 1
- LVDKZNITIUWNER-UHFFFAOYSA-N Bronopol Chemical compound OCC(Br)(CO)[N+]([O-])=O LVDKZNITIUWNER-UHFFFAOYSA-N 0.000 description 1
- CIUUIPMOFZIWIZ-UHFFFAOYSA-N Bropirimine Chemical compound NC1=NC(O)=C(Br)C(C=2C=CC=CC=2)=N1 CIUUIPMOFZIWIZ-UHFFFAOYSA-N 0.000 description 1
- UMSGKTJDUHERQW-UHFFFAOYSA-N Brotizolam Chemical compound C1=2C=C(Br)SC=2N2C(C)=NN=C2CN=C1C1=CC=CC=C1Cl UMSGKTJDUHERQW-UHFFFAOYSA-N 0.000 description 1
- IRQXZTBHNKVIRL-GOTQHHPNSA-N Bruceantin Chemical compound CC1=C(O)C(=O)C[C@]2(C)[C@@H]([C@@H](O)[C@@H]3O)[C@@]45CO[C@@]3(C(=O)OC)[C@@H]5[C@@H](OC(=O)\C=C(/C)C(C)C)C(=O)O[C@@H]4C[C@H]21 IRQXZTBHNKVIRL-GOTQHHPNSA-N 0.000 description 1
- LIAWQASKBFCRNR-UHFFFAOYSA-N Bucetin Chemical compound CCOC1=CC=C(NC(=O)CC(C)O)C=C1 LIAWQASKBFCRNR-UHFFFAOYSA-N 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- 229930190011 Bufogenin Natural products 0.000 description 1
- RHLJLALHBZGAFM-UHFFFAOYSA-N Bunazosinum Chemical compound C1CN(C(=O)CCC)CCCN1C1=NC(N)=C(C=C(OC)C(OC)=C2)C2=N1 RHLJLALHBZGAFM-UHFFFAOYSA-N 0.000 description 1
- VKSPIPWLHGKJQO-UHFFFAOYSA-N Bupicomide Chemical compound CCCCC1=CC=C(C(N)=O)N=C1 VKSPIPWLHGKJQO-UHFFFAOYSA-N 0.000 description 1
- WLYWZMQECNKDLI-UHFFFAOYSA-N Buterizine Chemical compound C=1C=C2N(CC)C(CCCC)=NC2=CC=1CN(CC1)CCN1C(C=1C=CC=CC=1)C1=CC=CC=C1 WLYWZMQECNKDLI-UHFFFAOYSA-N 0.000 description 1
- 229930183180 Butirosin Natural products 0.000 description 1
- BKAQXYNWONVOAX-UHFFFAOYSA-N Butonate Chemical compound CCCC(=O)OC(C(Cl)(Cl)Cl)P(=O)(OC)OC BKAQXYNWONVOAX-UHFFFAOYSA-N 0.000 description 1
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 1
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- MMGJNINGVUMRFI-KAOUJKNGSA-N C(C1CC1)N1CC[C@@]23CCCCC2[C@@H]1Cc1c3[nH]c2ccccc12 Chemical compound C(C1CC1)N1CC[C@@]23CCCCC2[C@@H]1Cc1c3[nH]c2ccccc12 MMGJNINGVUMRFI-KAOUJKNGSA-N 0.000 description 1
- BZUKDLNEDCDORL-ULRSWZSCSA-N C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(=O)NCN(C)CCN(C)CNC(=O)C=5C([C@@]6(O)C(O)=C7[C@@H]([C@](C8=CC=CC(O)=C8C7=O)(C)O)C[C@H]6[C@@H](C=5O)N(C)C)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O Chemical compound C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(=O)NCN(C)CCN(C)CNC(=O)C=5C([C@@]6(O)C(O)=C7[C@@H]([C@](C8=CC=CC(O)=C8C7=O)(C)O)C[C@H]6[C@@H](C=5O)N(C)C)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O BZUKDLNEDCDORL-ULRSWZSCSA-N 0.000 description 1
- NACJSVRGPPMZRS-BWUCMLAFSA-N CC(C)NCC(O)COc1ccccc1[C@H]1C[C@@H]2CC[C@H]1C2 Chemical compound CC(C)NCC(O)COc1ccccc1[C@H]1C[C@@H]2CC[C@H]1C2 NACJSVRGPPMZRS-BWUCMLAFSA-N 0.000 description 1
- KHFUQWURHSKTPO-LBYUQGKWSA-N CC(C)c1cc(\N=N\c2ccc(cc2)S(=O)(=O)c2ccc(cc2)\N=N\c2cc(C(C)C)c(O)cc2C)c(C)cc1O Chemical compound CC(C)c1cc(\N=N\c2ccc(cc2)S(=O)(=O)c2ccc(cc2)\N=N\c2cc(C(C)C)c(O)cc2C)c(C)cc1O KHFUQWURHSKTPO-LBYUQGKWSA-N 0.000 description 1
- COXVTLYNGOIATD-HVMBLDELSA-N CC1=C(C=CC(=C1)C1=CC(C)=C(C=C1)\N=N\C1=C(O)C2=C(N)C(=CC(=C2C=C1)S(O)(=O)=O)S(O)(=O)=O)\N=N\C1=CC=C2C(=CC(=C(N)C2=C1O)S(O)(=O)=O)S(O)(=O)=O Chemical compound CC1=C(C=CC(=C1)C1=CC(C)=C(C=C1)\N=N\C1=C(O)C2=C(N)C(=CC(=C2C=C1)S(O)(=O)=O)S(O)(=O)=O)\N=N\C1=CC=C2C(=CC(=C(N)C2=C1O)S(O)(=O)=O)S(O)(=O)=O COXVTLYNGOIATD-HVMBLDELSA-N 0.000 description 1
- NXAWWUAMFJNVMF-FELNFDIXSA-M CC[C@@H]1[C@H]([N@+]2([C@H]3CC1[C@@H]4[C@@H]2C[C@@]5([C@H]3N(C6=CC=CC=C65)C)[C@@H]4O)CC(CN(CC)CC)O)O.[C@@H]([C@H](C(=O)[O-])O)(C(=O)O)O Chemical compound CC[C@@H]1[C@H]([N@+]2([C@H]3CC1[C@@H]4[C@@H]2C[C@@]5([C@H]3N(C6=CC=CC=C65)C)[C@@H]4O)CC(CN(CC)CC)O)O.[C@@H]([C@H](C(=O)[O-])O)(C(=O)O)O NXAWWUAMFJNVMF-FELNFDIXSA-M 0.000 description 1
- 108010058905 CD44v6 antigen Proteins 0.000 description 1
- RZZPDXZPRHQOCG-OJAKKHQRSA-M CDP-choline(1-) Chemical compound O[C@@H]1[C@H](O)[C@@H](COP([O-])(=O)OP([O-])(=O)OCC[N+](C)(C)C)O[C@H]1N1C(=O)N=C(N)C=C1 RZZPDXZPRHQOCG-OJAKKHQRSA-M 0.000 description 1
- XZPAMMPYTOAFOU-MUQADHOPSA-N CN1[C@@H]2CC[C@H]1C[C@H](C2)OC(=O)[C@](C)(CO)c3ccccc3 Chemical compound CN1[C@@H]2CC[C@H]1C[C@H](C2)OC(=O)[C@](C)(CO)c3ccccc3 XZPAMMPYTOAFOU-MUQADHOPSA-N 0.000 description 1
- KORNTPPJEAJQIU-KJXAQDMKSA-N Cabaser Chemical compound C1=CC([C@H]2C[C@H](CN(CC=C)[C@@H]2C2)C(=O)N(CCCN(C)C)C(=O)NCC)=C3C2=CNC3=C1 KORNTPPJEAJQIU-KJXAQDMKSA-N 0.000 description 1
- QLTVVOATEHFXLT-UHFFFAOYSA-N Cadralazine Chemical compound CCOC(=O)NNC1=CC=C(N(CC)CC(C)O)N=N1 QLTVVOATEHFXLT-UHFFFAOYSA-N 0.000 description 1
- 235000021318 Calcifediol Nutrition 0.000 description 1
- 239000001736 Calcium glycerylphosphate Substances 0.000 description 1
- 239000002970 Calcium lactobionate Substances 0.000 description 1
- 102100033620 Calponin-1 Human genes 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- GHOSNRCGJFBJIB-UHFFFAOYSA-N Candesartan cilexetil Chemical compound C=12N(CC=3C=CC(=CC=3)C=3C(=CC=CC=3)C3=NNN=N3)C(OCC)=NC2=CC=CC=1C(=O)OC(C)OC(=O)OC1CCCCC1 GHOSNRCGJFBJIB-UHFFFAOYSA-N 0.000 description 1
- VBGLYOIFKLUMQG-UHFFFAOYSA-N Cannabinol Chemical compound C1=C(C)C=C2C3=C(O)C=C(CCCCC)C=C3OC(C)(C)C2=C1 VBGLYOIFKLUMQG-UHFFFAOYSA-N 0.000 description 1
- VJKVHUZOVAWKEB-UHFFFAOYSA-N Carbestrol Chemical compound C1CC(C(O)=O)C(C)C(CC)=C1C1=CC=C(OC)C=C1 VJKVHUZOVAWKEB-UHFFFAOYSA-N 0.000 description 1
- 229930188120 Carbomycin Natural products 0.000 description 1
- VEDTXTNSFWUXGQ-UHFFFAOYSA-N Carbophenothion Chemical compound CCOP(=S)(OCC)SCSC1=CC=C(Cl)C=C1 VEDTXTNSFWUXGQ-UHFFFAOYSA-N 0.000 description 1
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- OVEVHVURWWTPFC-UHFFFAOYSA-N Carnidazole Chemical compound COC(=S)NCCN1C(C)=NC=C1[N+]([O-])=O OVEVHVURWWTPFC-UHFFFAOYSA-N 0.000 description 1
- 102000005600 Cathepsins Human genes 0.000 description 1
- 108010084457 Cathepsins Proteins 0.000 description 1
- 102100032231 Caveolae-associated protein 2 Human genes 0.000 description 1
- 108050005259 Caveolae-associated protein 2 Proteins 0.000 description 1
- UQLLWWBDSUHNEB-CZUORRHYSA-N Cefaprin Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC(=O)C)C(O)=O)C(=O)CSC1=CC=NC=C1 UQLLWWBDSUHNEB-CZUORRHYSA-N 0.000 description 1
- QYQDKDWGWDOFFU-IUODEOHRSA-N Cefotiam Chemical compound CN(C)CCN1N=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)CC=3N=C(N)SC=3)[C@H]2SC1 QYQDKDWGWDOFFU-IUODEOHRSA-N 0.000 description 1
- JOATXPAWOHTVSZ-UHFFFAOYSA-N Celiprolol Chemical compound CCN(CC)C(=O)NC1=CC=C(OCC(O)CNC(C)(C)C)C(C(C)=O)=C1 JOATXPAWOHTVSZ-UHFFFAOYSA-N 0.000 description 1
- MMNICIJVQJJHHF-UHFFFAOYSA-N Cetiedil Chemical compound C1CCCCC1C(C1=CSC=C1)C(=O)OCCN1CCCCCC1 MMNICIJVQJJHHF-UHFFFAOYSA-N 0.000 description 1
- ZKLPARSLTMPFCP-UHFFFAOYSA-N Cetirizine Chemical compound C1CN(CCOCC(=O)O)CCN1C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 ZKLPARSLTMPFCP-UHFFFAOYSA-N 0.000 description 1
- NPBVQXIMTZKSBA-UHFFFAOYSA-N Chavibetol Natural products COC1=CC=C(CC=C)C=C1O NPBVQXIMTZKSBA-UHFFFAOYSA-N 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- QDHHCQZDFGDHMP-UHFFFAOYSA-N Chloramine Chemical compound ClN QDHHCQZDFGDHMP-UHFFFAOYSA-N 0.000 description 1
- LIRCDOVJWUGTMW-ZWNOBZJWSA-N Chloramphenicol succinate Chemical compound OC(=O)CCC(=O)OC[C@@H](NC(=O)C(Cl)Cl)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 LIRCDOVJWUGTMW-ZWNOBZJWSA-N 0.000 description 1
- GHXZTYHSJHQHIJ-UHFFFAOYSA-N Chlorhexidine Chemical compound C=1C=C(Cl)C=CC=1NC(N)=NC(N)=NCCCCCCN=C(N)N=C(N)NC1=CC=C(Cl)C=C1 GHXZTYHSJHQHIJ-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- QMBJSIBWORFWQT-DFXBJWIESA-N Chlormadinone acetate Chemical compound C1=C(Cl)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 QMBJSIBWORFWQT-DFXBJWIESA-N 0.000 description 1
- PCLITLDOTJTVDJ-UHFFFAOYSA-N Chlormethiazole Chemical compound CC=1N=CSC=1CCCl PCLITLDOTJTVDJ-UHFFFAOYSA-N 0.000 description 1
- VOPWNXZWBYDODV-UHFFFAOYSA-N Chlorodifluoromethane Chemical compound FC(F)Cl VOPWNXZWBYDODV-UHFFFAOYSA-N 0.000 description 1
- 239000004340 Chloropentafluoroethane Substances 0.000 description 1
- RKWGIWYCVPQPMF-UHFFFAOYSA-N Chloropropamide Chemical compound CCCNC(=O)NS(=O)(=O)C1=CC=C(Cl)C=C1 RKWGIWYCVPQPMF-UHFFFAOYSA-N 0.000 description 1
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 1
- MKQWTWSXVILIKJ-LXGUWJNJSA-N Chlorozotocin Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](C=O)NC(=O)N(N=O)CCCl MKQWTWSXVILIKJ-LXGUWJNJSA-N 0.000 description 1
- ZCKAMNXUHHNZLN-UHFFFAOYSA-N Chlorphentermine Chemical compound CC(C)(N)CC1=CC=C(Cl)C=C1 ZCKAMNXUHHNZLN-UHFFFAOYSA-N 0.000 description 1
- 239000004099 Chlortetracycline Substances 0.000 description 1
- 101800001982 Cholecystokinin Proteins 0.000 description 1
- 102100025841 Cholecystokinin Human genes 0.000 description 1
- 239000004380 Cholic acid Substances 0.000 description 1
- 235000019743 Choline chloride Nutrition 0.000 description 1
- 108010009685 Cholinergic Receptors Proteins 0.000 description 1
- IPOBOOXFSRWSHL-UHFFFAOYSA-N Cibenzoline Chemical compound C=1C=CC=CC=1C1(C=2C=CC=CC=2)CC1C1=NCCN1 IPOBOOXFSRWSHL-UHFFFAOYSA-N 0.000 description 1
- CVKNDPRBJVBDSS-UHFFFAOYSA-N Cicletanine Chemical compound O1CC2=C(O)C(C)=NC=C2C1C1=CC=C(Cl)C=C1 CVKNDPRBJVBDSS-UHFFFAOYSA-N 0.000 description 1
- UVAUYSRYXACKSC-ULQDDVLXSA-N Cilazaprilat Chemical compound C([C@@H](C(=O)O)N[C@@H]1C(N2[C@@H](CCCN2CCC1)C(O)=O)=O)CC1=CC=CC=C1 UVAUYSRYXACKSC-ULQDDVLXSA-N 0.000 description 1
- 235000001258 Cinchona calisaya Nutrition 0.000 description 1
- VCMZUKHJKLNFPH-JXMROGBWSA-N Cinperene Chemical compound O=C1NC(=O)CCC1(C=1C=CC=CC=1)C1CCN(C\C=C\C=2C=CC=CC=2)CC1 VCMZUKHJKLNFPH-JXMROGBWSA-N 0.000 description 1
- KATBVKFXGKGUFE-UHFFFAOYSA-N Cintazone Chemical compound C12=CC=CC=C2N2C(=O)C(CCCCC)C(=O)N2C=C1C1=CC=CC=C1 KATBVKFXGKGUFE-UHFFFAOYSA-N 0.000 description 1
- KPSRODZRAIWAKH-JTQLQIEISA-N Ciprofibrate Natural products C1=CC(OC(C)(C)C(O)=O)=CC=C1[C@H]1C(Cl)(Cl)C1 KPSRODZRAIWAKH-JTQLQIEISA-N 0.000 description 1
- VKPYUUBEDXIQIB-QBPWRKFFSA-N Ciprostene Chemical compound C1\C(=C/CCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@]21C VKPYUUBEDXIQIB-QBPWRKFFSA-N 0.000 description 1
- YAORIDZYZDUZCM-UHFFFAOYSA-N Cirazoline Chemical compound N=1CCNC=1COC1=CC=CC=C1C1CC1 YAORIDZYZDUZCM-UHFFFAOYSA-N 0.000 description 1
- DTCHCUZKLHCAAF-AOBHSWNLSA-N ClC[C@@]12C(=C(C[C@H]1[C@@H]1CC[C@H]3CC(=O)CC[C@]3(C)[C@H]1CC2)O)O Chemical compound ClC[C@@]12C(=C(C[C@H]1[C@@H]1CC[C@H]3CC(=O)CC[C@]3(C)[C@H]1CC2)O)O DTCHCUZKLHCAAF-AOBHSWNLSA-N 0.000 description 1
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 description 1
- TUQAUZURSHKTFF-UHFFFAOYSA-N Clazolimine Chemical compound N=C1N(C)C(=O)CN1C1=CC=C(Cl)C=C1 TUQAUZURSHKTFF-UHFFFAOYSA-N 0.000 description 1
- OIRAEJWYWSAQNG-UHFFFAOYSA-N Clidanac Chemical compound ClC=1C=C2C(C(=O)O)CCC2=CC=1C1CCCCC1 OIRAEJWYWSAQNG-UHFFFAOYSA-N 0.000 description 1
- BMOVQUBVGICXQN-UHFFFAOYSA-N Clinofibrate Chemical compound C1=CC(OC(C)(CC)C(O)=O)=CC=C1C1(C=2C=CC(OC(C)(CC)C(O)=O)=CC=2)CCCCC1 BMOVQUBVGICXQN-UHFFFAOYSA-N 0.000 description 1
- QCDFBFJGMNKBDO-UHFFFAOYSA-N Clioquinol Chemical compound C1=CN=C2C(O)=C(I)C=C(Cl)C2=C1 QCDFBFJGMNKBDO-UHFFFAOYSA-N 0.000 description 1
- ARPLCFGLEYFDCN-CDACMRRYSA-N Clocortolone acetate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(Cl)[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)COC(C)=O)[C@@]2(C)C[C@@H]1O ARPLCFGLEYFDCN-CDACMRRYSA-N 0.000 description 1
- NENBAISIHCWPKP-UHFFFAOYSA-N Clofenamide Chemical compound NS(=O)(=O)C1=CC=C(Cl)C(S(N)(=O)=O)=C1 NENBAISIHCWPKP-UHFFFAOYSA-N 0.000 description 1
- UMRURYMAPMZKQO-NDKKBYRMSA-N Clometherone Chemical compound C1([C@@H](Cl)C2)=CC(=O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(C)=O)[C@@]2(C)CC1 UMRURYMAPMZKQO-NDKKBYRMSA-N 0.000 description 1
- GDLIGKIOYRNHDA-UHFFFAOYSA-N Clomipramine Chemical compound C1CC2=CC=C(Cl)C=C2N(CCCN(C)C)C2=CC=CC=C21 GDLIGKIOYRNHDA-UHFFFAOYSA-N 0.000 description 1
- GJSURZIOUXUGAL-UHFFFAOYSA-N Clonidine Chemical compound ClC1=CC=CC(Cl)=C1NC1=NCCN1 GJSURZIOUXUGAL-UHFFFAOYSA-N 0.000 description 1
- ZDPIZLCVJAAHHR-UHFFFAOYSA-N Clopidol Chemical compound CC1=NC(C)=C(Cl)C(O)=C1Cl ZDPIZLCVJAAHHR-UHFFFAOYSA-N 0.000 description 1
- VJGGHXVGBSZVMZ-QIZQQNKQSA-N Cloprostenol Chemical compound C([C@H](O)\C=C\[C@@H]1[C@H]([C@@H](O)C[C@H]1O)C\C=C/CCCC(O)=O)OC1=CC=CC(Cl)=C1 VJGGHXVGBSZVMZ-QIZQQNKQSA-N 0.000 description 1
- VPMWFZKOWULPGT-UHFFFAOYSA-N Clorexolone Chemical compound C1C=2C=C(Cl)C(S(=O)(=O)N)=CC=2C(=O)N1C1CCCCC1 VPMWFZKOWULPGT-UHFFFAOYSA-N 0.000 description 1
- KAAZGXDPUNNEFN-UHFFFAOYSA-N Clotiapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2SC2=CC=C(Cl)C=C12 KAAZGXDPUNNEFN-UHFFFAOYSA-N 0.000 description 1
- CHBRHODLKOZEPZ-UHFFFAOYSA-N Clotiazepam Chemical compound S1C(CC)=CC2=C1N(C)C(=O)CN=C2C1=CC=CC=C1Cl CHBRHODLKOZEPZ-UHFFFAOYSA-N 0.000 description 1
- 102100023804 Coagulation factor VII Human genes 0.000 description 1
- 108010048623 Collagen Receptors Proteins 0.000 description 1
- 102000012422 Collagen Type I Human genes 0.000 description 1
- 108010022452 Collagen Type I Proteins 0.000 description 1
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 1
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 1
- AYYIATOVDZJSNR-UHFFFAOYSA-N Conessine Natural products CC1C2CCC3C4(C)CC=C5CC(CCC5(C)C4CCC23CN1C)N(C)C AYYIATOVDZJSNR-UHFFFAOYSA-N 0.000 description 1
- OCUCCJIRFHNWBP-IYEMJOQQSA-L Copper gluconate Chemical compound [Cu+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O OCUCCJIRFHNWBP-IYEMJOQQSA-L 0.000 description 1
- YXKFATPOEMHNMJ-KJEYTGHBSA-N Cormethasone acetate Chemical compound C1C(F)(F)C2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)COC(C)=O)(O)[C@@]1(C)C[C@@H]2O YXKFATPOEMHNMJ-KJEYTGHBSA-N 0.000 description 1
- OMFXVFTZEKFJBZ-UHFFFAOYSA-N Corticosterone Natural products O=C1CCC2(C)C3C(O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 OMFXVFTZEKFJBZ-UHFFFAOYSA-N 0.000 description 1
- UIKROCXWUNQSPJ-UHFFFAOYSA-N Cotinine Natural products C1CC(=O)N(C)C1C1=CC=CN=C1 UIKROCXWUNQSPJ-UHFFFAOYSA-N 0.000 description 1
- 240000004090 Crataegus azarolus Species 0.000 description 1
- 235000013176 Crataegus azarolus Nutrition 0.000 description 1
- WHPAGCJNPTUGGD-UHFFFAOYSA-N Croconazole Chemical compound ClC1=CC=CC(COC=2C(=CC=CC=2)C(=C)N2C=NC=C2)=C1 WHPAGCJNPTUGGD-UHFFFAOYSA-N 0.000 description 1
- TVZCRIROJQEVOT-CABCVRRESA-N Cromakalim Chemical compound N1([C@@H]2C3=CC(=CC=C3OC([C@H]2O)(C)C)C#N)CCCC1=O TVZCRIROJQEVOT-CABCVRRESA-N 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- BOFHKBLZOYVHSI-UHFFFAOYSA-N Crufomate Chemical compound CNP(=O)(OC)OC1=CC=C(C(C)(C)C)C=C1Cl BOFHKBLZOYVHSI-UHFFFAOYSA-N 0.000 description 1
- 229920001651 Cyanoacrylate Polymers 0.000 description 1
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 1
- GVOUFPWUYJWQSK-UHFFFAOYSA-N Cyclofenil Chemical compound C1=CC(OC(=O)C)=CC=C1C(C=1C=CC(OC(C)=O)=CC=1)=C1CCCCC1 GVOUFPWUYJWQSK-UHFFFAOYSA-N 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- SITQVDJAXQSXSA-CEZRHVESSA-N Cynarin Natural products O[C@@H]1C[C@@](C[C@H](O)[C@H]1OC(=O)C=Cc2ccc(O)c(O)c2)(OC(=O)C=Cc3cccc(O)c3O)C(=O)O SITQVDJAXQSXSA-CEZRHVESSA-N 0.000 description 1
- YDDUMTOHNYZQPO-RVXRWRFUSA-N Cynarine Chemical compound O([C@@H]1C[C@@](C[C@H]([C@@H]1O)O)(OC(=O)\C=C\C=1C=C(O)C(O)=CC=1)C(O)=O)C(=O)\C=C\C1=CC=C(O)C(O)=C1 YDDUMTOHNYZQPO-RVXRWRFUSA-N 0.000 description 1
- DYDCUQKUCUHJBH-UWTATZPHSA-N D-Cycloserine Chemical compound N[C@@H]1CONC1=O DYDCUQKUCUHJBH-UWTATZPHSA-N 0.000 description 1
- DYDCUQKUCUHJBH-UHFFFAOYSA-N D-Cycloserine Natural products NC1CONC1=O DYDCUQKUCUHJBH-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- SPKNARKFCOPTSY-UHFFFAOYSA-N D-asperlin Natural products CC1OC1C1C(OC(C)=O)C=CC(=O)O1 SPKNARKFCOPTSY-UHFFFAOYSA-N 0.000 description 1
- 239000011665 D-biotin Substances 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- UYUXSRADSPPKRZ-SKNVOMKLSA-N D-glucurono-6,3-lactone Chemical compound O=C[C@H](O)[C@H]1OC(=O)[C@@H](O)[C@H]1O UYUXSRADSPPKRZ-SKNVOMKLSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine group Chemical group N[C@H](CCCCN)C(=O)O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- SNPLKNRPJHDVJA-ZETCQYMHSA-N D-panthenol Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCCO SNPLKNRPJHDVJA-ZETCQYMHSA-N 0.000 description 1
- 235000004866 D-panthenol Nutrition 0.000 description 1
- 239000011703 D-panthenol Substances 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 1
- YVGGHNCTFXOJCH-UHFFFAOYSA-N DDT Chemical compound C1=CC(Cl)=CC=C1C(C(Cl)(Cl)Cl)C1=CC=C(Cl)C=C1 YVGGHNCTFXOJCH-UHFFFAOYSA-N 0.000 description 1
- 102000052510 DNA-Binding Proteins Human genes 0.000 description 1
- 108700020911 DNA-Binding Proteins Proteins 0.000 description 1
- 229920004934 Dacron® Polymers 0.000 description 1
- 108010013198 Daptomycin Proteins 0.000 description 1
- HFPMXDMZJUJZBX-AWEZNQCLSA-N Deacetylcolchicine Chemical compound C1([C@@H](N)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC HFPMXDMZJUJZBX-AWEZNQCLSA-N 0.000 description 1
- 108010000437 Deamino Arginine Vasopressin Proteins 0.000 description 1
- XMSXQFUHVRWGNA-UHFFFAOYSA-N Decamethylcyclopentasiloxane Chemical compound C[Si]1(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O1 XMSXQFUHVRWGNA-UHFFFAOYSA-N 0.000 description 1
- 102000004237 Decorin Human genes 0.000 description 1
- 108090000738 Decorin Proteins 0.000 description 1
- 239000004287 Dehydroacetic acid Substances 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- FMTDIUIBLCQGJB-UHFFFAOYSA-N Demethylchlortetracyclin Natural products C1C2C(O)C3=C(Cl)C=CC(O)=C3C(=O)C2=C(O)C2(O)C1C(N(C)C)C(O)=C(C(N)=O)C2=O FMTDIUIBLCQGJB-UHFFFAOYSA-N 0.000 description 1
- VPGRYOFKCNULNK-ACXQXYJUSA-N Deoxycorticosterone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)COC(=O)C)[C@@]1(C)CC2 VPGRYOFKCNULNK-ACXQXYJUSA-N 0.000 description 1
- BSHYASCHOGHGHW-PIQRJGQMSA-N Descinolone acetonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)C)[C@@]1(C)C[C@@H]2O BSHYASCHOGHGHW-PIQRJGQMSA-N 0.000 description 1
- CVBMAZKKCSYWQR-BPJCFPRXSA-N Deserpidine Natural products O=C(OC)[C@@H]1[C@H](OC)[C@H](OC(=O)c2cc(OC)c(OC)c(OC)c2)C[C@H]2[C@@H]1C[C@H]1N(C2)CCc2c3c([nH]c12)cccc3 CVBMAZKKCSYWQR-BPJCFPRXSA-N 0.000 description 1
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 description 1
- 239000012848 Dextrorphan Substances 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- AUGQEEXBDZWUJY-ZLJUKNTDSA-N Diacetoxyscirpenol Chemical compound C([C@]12[C@]3(C)[C@H](OC(C)=O)[C@@H](O)[C@H]1O[C@@H]1C=C(C)CC[C@@]13COC(=O)C)O2 AUGQEEXBDZWUJY-ZLJUKNTDSA-N 0.000 description 1
- AUGQEEXBDZWUJY-UHFFFAOYSA-N Diacetoxyscirpenol Natural products CC(=O)OCC12CCC(C)=CC1OC1C(O)C(OC(C)=O)C2(C)C11CO1 AUGQEEXBDZWUJY-UHFFFAOYSA-N 0.000 description 1
- AZJUFRDUYTYIHV-NKFKGCMQSA-N Dibenzoyl Thiamine Chemical compound C=1C=CC=CC=1C(=O)OCC\C(SC(=O)C=1C=CC=CC=1)=C(/C)N(C=O)CC1=CN=C(C)N=C1N AZJUFRDUYTYIHV-NKFKGCMQSA-N 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- MDNWOSOZYLHTCG-UHFFFAOYSA-N Dichlorophen Chemical compound OC1=CC=C(Cl)C=C1CC1=CC(Cl)=CC=C1O MDNWOSOZYLHTCG-UHFFFAOYSA-N 0.000 description 1
- WUVPAYPBMZMHJO-IMNLCBETSA-N Dicirenone Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)C(=O)OC(C)C)C[C@@]21CCC(=O)O1 WUVPAYPBMZMHJO-IMNLCBETSA-N 0.000 description 1
- PHHROXLDZHUIGO-PNBTUHDLSA-N Didrovaltratum Chemical compound C([C@@]12[C@@H](OC(C)=O)C[C@H]3[C@@H]1[C@H](OC(=O)CC(C)C)OC=C3COC(=O)CC(C)C)O2 PHHROXLDZHUIGO-PNBTUHDLSA-N 0.000 description 1
- OEHKWMHRFWWMQK-UHFFFAOYSA-N Difeterol Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)OCCN(C)C(C)C(O)C1=CC=CC=C1 OEHKWMHRFWWMQK-UHFFFAOYSA-N 0.000 description 1
- WYQPLTPSGFELIB-JTQPXKBDSA-N Difluprednate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2CC[C@@](C(=O)COC(C)=O)(OC(=O)CCC)[C@@]2(C)C[C@@H]1O WYQPLTPSGFELIB-JTQPXKBDSA-N 0.000 description 1
- CKNOLMVLQUPVMU-XOMFLMSUSA-N Digitalin Natural products O(C)[C@H]1[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@@H](O)[C@H](CO)O2)[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@@](C)([C@@H]([C@H](O)C5)C5=CC(=O)OC5)CC4)CC3)CC2)[C@@H]1O CKNOLMVLQUPVMU-XOMFLMSUSA-N 0.000 description 1
- FWCBATIDXGJRMF-FLNNQWSLSA-N Dikegulac Chemical compound O([C@H]12)C(C)(C)OC[C@@H]1O[C@]1(C(O)=O)[C@H]2OC(C)(C)O1 FWCBATIDXGJRMF-FLNNQWSLSA-N 0.000 description 1
- LVHOURKCKUYIGK-RGUJTQARSA-N Dimethisterone Chemical compound C1([C@@H](C)C2)=CC(=O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@](C#CC)(O)[C@@]2(C)CC1 LVHOURKCKUYIGK-RGUJTQARSA-N 0.000 description 1
- XPINIPXARSNZDM-VAWYXSNFSA-N Dimethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/C)=C(\C)C1=CC=C(O)C=C1 XPINIPXARSNZDM-VAWYXSNFSA-N 0.000 description 1
- ZEFNOZRLAWVAQF-UHFFFAOYSA-N Dinitolmide Chemical compound CC1=C(C(N)=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O ZEFNOZRLAWVAQF-UHFFFAOYSA-N 0.000 description 1
- MMDWBZHWHFGVHF-UHFFFAOYSA-N Dinsed Chemical compound [O-][N+](=O)C1=CC=CC(S(=O)(=O)NCCNS(=O)(=O)C=2C=C(C=CC=2)[N+]([O-])=O)=C1 MMDWBZHWHFGVHF-UHFFFAOYSA-N 0.000 description 1
- 241000551547 Dione <red algae> Species 0.000 description 1
- JYGLAHSAISAEAL-UHFFFAOYSA-N Diphenadione Chemical compound O=C1C2=CC=CC=C2C(=O)C1C(=O)C(C=1C=CC=CC=1)C1=CC=CC=C1 JYGLAHSAISAEAL-UHFFFAOYSA-N 0.000 description 1
- ADZXDFALMZGPTN-UHFFFAOYSA-M Diponium bromide Chemical compound [Br-].C1CCCC1C(C(=O)OCC[N+](CC)(CC)CC)C1CCCC1 ADZXDFALMZGPTN-UHFFFAOYSA-M 0.000 description 1
- OIJXLIIMXHRJJH-KNLIIKEYSA-N Diprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)C(C)(C)O)OC)CN2CC1CC1 OIJXLIIMXHRJJH-KNLIIKEYSA-N 0.000 description 1
- 229940123907 Disease modifying antirheumatic drug Drugs 0.000 description 1
- FVIGODVHAVLZOO-UHFFFAOYSA-N Dixanthogen Chemical compound CCOC(=S)SSC(=S)OCC FVIGODVHAVLZOO-UHFFFAOYSA-N 0.000 description 1
- JRWZLRBJNMZMFE-UHFFFAOYSA-N Dobutamine Chemical compound C=1C=C(O)C(O)=CC=1CCNC(C)CCC1=CC=C(O)C=C1 JRWZLRBJNMZMFE-UHFFFAOYSA-N 0.000 description 1
- ZLVMAMIPILWYHQ-INIZCTEOSA-N Docarpamine Chemical compound CCOC(=O)OC1=CC=C(CCNC(=O)[C@H](CCSC)NC(C)=O)C=C1OC(=O)OCC ZLVMAMIPILWYHQ-INIZCTEOSA-N 0.000 description 1
- GNZHVEIGGFMLSP-OZXSUGGESA-N Doconazole Chemical compound ClC1=CC(Cl)=CC=C1[C@]1(CN2C=NC=C2)O[C@@H](COC=2C=CC(=CC=2)C=2C=CC=CC=2)CO1 GNZHVEIGGFMLSP-OZXSUGGESA-N 0.000 description 1
- OJIYIVCMRYCWSE-UHFFFAOYSA-M Domiphen bromide Chemical compound [Br-].CCCCCCCCCCCC[N+](C)(C)CCOC1=CC=CC=C1 OJIYIVCMRYCWSE-UHFFFAOYSA-M 0.000 description 1
- VOJLELRQLPENHL-UHFFFAOYSA-N Doxefazepam Chemical compound N=1C(O)C(=O)N(CCO)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F VOJLELRQLPENHL-UHFFFAOYSA-N 0.000 description 1
- MWWSFMDVAYGXBV-RUELKSSGSA-N Doxorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-RUELKSSGSA-N 0.000 description 1
- ZQZFYGIXNQKOAV-OCEACIFDSA-N Droloxifene Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=C(O)C=CC=1)\C1=CC=C(OCCN(C)C)C=C1 ZQZFYGIXNQKOAV-OCEACIFDSA-N 0.000 description 1
- CYQFCXCEBYINGO-DLBZAZTESA-N Dronabinol Natural products C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@H]21 CYQFCXCEBYINGO-DLBZAZTESA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 1
- 102000001301 EGF receptor Human genes 0.000 description 1
- 108060006698 EGF receptor Proteins 0.000 description 1
- DYEFUKCXAQOFHX-UHFFFAOYSA-N Ebselen Chemical compound [se]1C2=CC=CC=C2C(=O)N1C1=CC=CC=C1 DYEFUKCXAQOFHX-UHFFFAOYSA-N 0.000 description 1
- ZVXBAHLOGZCFTP-UHFFFAOYSA-N Efloxate Chemical compound C=1C(OCC(=O)OCC)=CC=C(C(C=2)=O)C=1OC=2C1=CC=CC=C1 ZVXBAHLOGZCFTP-UHFFFAOYSA-N 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- AFSDNFLWKVMVRB-UHFFFAOYSA-N Ellagic acid Chemical compound OC1=C(O)C(OC2=O)=C3C4=C2C=C(O)C(O)=C4OC(=O)C3=C1 AFSDNFLWKVMVRB-UHFFFAOYSA-N 0.000 description 1
- ATJXMQHAMYVHRX-CPCISQLKSA-N Ellagic acid Natural products OC1=C(O)[C@H]2OC(=O)c3cc(O)c(O)c4OC(=O)C(=C1)[C@H]2c34 ATJXMQHAMYVHRX-CPCISQLKSA-N 0.000 description 1
- 229920002079 Ellagic acid Polymers 0.000 description 1
- MBYXEBXZARTUSS-QLWBXOBMSA-N Emetamine Natural products O(C)c1c(OC)cc2c(c(C[C@@H]3[C@H](CC)CN4[C@H](c5c(cc(OC)c(OC)c5)CC4)C3)ncc2)c1 MBYXEBXZARTUSS-QLWBXOBMSA-N 0.000 description 1
- URJQOOISAKEBKW-UHFFFAOYSA-N Emorfazone Chemical compound C1=NN(C)C(=O)C(OCC)=C1N1CCOCC1 URJQOOISAKEBKW-UHFFFAOYSA-N 0.000 description 1
- 108010066671 Enalaprilat Proteins 0.000 description 1
- 102100037241 Endoglin Human genes 0.000 description 1
- 108010036395 Endoglin Proteins 0.000 description 1
- 102100038083 Endosialin Human genes 0.000 description 1
- 101710144543 Endosialin Proteins 0.000 description 1
- 102400001047 Endostatin Human genes 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- 108010041308 Endothelial Growth Factors Proteins 0.000 description 1
- 102000010180 Endothelin receptor Human genes 0.000 description 1
- 108050001739 Endothelin receptor Proteins 0.000 description 1
- SAMRUMKYXPVKPA-VFKOLLTISA-N Enocitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 SAMRUMKYXPVKPA-VFKOLLTISA-N 0.000 description 1
- NBEALWAVEGMZQY-UHFFFAOYSA-N Enpromate Chemical compound C=1C=CC=CC=1C(C#C)(C=1C=CC=CC=1)OC(=O)NC1CCCCC1 NBEALWAVEGMZQY-UHFFFAOYSA-N 0.000 description 1
- 101710204837 Envelope small membrane protein Proteins 0.000 description 1
- 108010038532 Enviomycin Proteins 0.000 description 1
- YARKMNAWFIMDKV-UHFFFAOYSA-N Epanolol Chemical compound C=1C=CC=C(C#N)C=1OCC(O)CNCCNC(=O)CC1=CC=C(O)C=C1 YARKMNAWFIMDKV-UHFFFAOYSA-N 0.000 description 1
- LMXFPUVUUSHCMM-UHFFFAOYSA-N Epicainide Chemical compound CCN1CCCC1CNC(=O)C(O)(C=1C=CC=CC=1)C1=CC=CC=C1 LMXFPUVUUSHCMM-UHFFFAOYSA-N 0.000 description 1
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 1
- UHQGCIIQUZBJAE-RRQVMCLOSA-N Epimestrol Chemical compound C1C[C@]2(C)[C@H](O)[C@H](O)C[C@H]2[C@@H]2CCC3=CC(OC)=CC=C3[C@H]21 UHQGCIIQUZBJAE-RRQVMCLOSA-N 0.000 description 1
- RHAXSHUQNIEUEY-UHFFFAOYSA-N Epirizole Chemical compound COC1=CC(C)=NN1C1=NC(C)=CC(OC)=N1 RHAXSHUQNIEUEY-UHFFFAOYSA-N 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- OBMLHUPNRURLOK-XGRAFVIBSA-N Epitiostanol Chemical compound C1[C@@H]2S[C@@H]2C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 OBMLHUPNRURLOK-XGRAFVIBSA-N 0.000 description 1
- WKRLQDKEXYKHJB-UHFFFAOYSA-N Equilin Natural products OC1=CC=C2C3CCC(C)(C(CC4)=O)C4C3=CCC2=C1 WKRLQDKEXYKHJB-UHFFFAOYSA-N 0.000 description 1
- WVVSZNPYNCNODU-CJBNDPTMSA-N Ergometrine Natural products C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@@H](CO)C)C2)=C3C2=CNC3=C1 WVVSZNPYNCNODU-CJBNDPTMSA-N 0.000 description 1
- DNVPQKQSNYMLRS-NXVQYWJNSA-N Ergosterol Natural products CC(C)[C@@H](C)C=C[C@H](C)[C@H]1CC[C@H]2C3=CC=C4C[C@@H](O)CC[C@]4(C)[C@@H]3CC[C@]12C DNVPQKQSNYMLRS-NXVQYWJNSA-N 0.000 description 1
- CNBHDDBNEKKMJH-ZFWWWQNUSA-N Eseridine Chemical compound O1N(C)CC[C@@]2(C)C3=CC(OC(=O)NC)=CC=C3N(C)[C@H]21 CNBHDDBNEKKMJH-ZFWWWQNUSA-N 0.000 description 1
- CNBHDDBNEKKMJH-UHFFFAOYSA-N Eseridine Natural products O1N(C)CCC2(C)C3=CC(OC(=O)NC)=CC=C3N(C)C21 CNBHDDBNEKKMJH-UHFFFAOYSA-N 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- UOACKFBJUYNSLK-XRKIENNPSA-N Estradiol Cypionate Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H](C4=CC=C(O)C=C4CC3)CC[C@@]21C)C(=O)CCC1CCCC1 UOACKFBJUYNSLK-XRKIENNPSA-N 0.000 description 1
- JQIYNMYZKRGDFK-RUFWAXPRSA-N Estradiol dipropionate Chemical compound C1CC2=CC(OC(=O)CC)=CC=C2[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CC)[C@@]1(C)CC2 JQIYNMYZKRGDFK-RUFWAXPRSA-N 0.000 description 1
- TXHUMRBWIWWBGW-UHFFFAOYSA-N Estradiol undecylate Natural products C1CC2=CC(O)=CC=C2C2C1C1CCC(OC(=O)CCCCCCCCCC)C1(C)CC2 TXHUMRBWIWWBGW-UHFFFAOYSA-N 0.000 description 1
- RSEPBGGWRJCQGY-RBRWEJTLSA-N Estradiol valerate Chemical compound C1CC2=CC(O)=CC=C2[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CCCC)[C@@]1(C)CC2 RSEPBGGWRJCQGY-RBRWEJTLSA-N 0.000 description 1
- DNXHEGUUPJUMQT-CBZIJGRNSA-N Estrone Chemical compound OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 DNXHEGUUPJUMQT-CBZIJGRNSA-N 0.000 description 1
- HBGOLJKPSFNJSD-UHFFFAOYSA-N Etamsylate Chemical compound CC[NH2+]CC.OC1=CC=C(O)C(S([O-])(=O)=O)=C1 HBGOLJKPSFNJSD-UHFFFAOYSA-N 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- KGWDUNBJIMUFAP-KVVVOXFISA-N Ethanolamine Oleate Chemical compound NCCO.CCCCCCCC\C=C/CCCCCCCC(O)=O KGWDUNBJIMUFAP-KVVVOXFISA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 1
- CUCHJCMWNFEYOM-UHFFFAOYSA-N Ethyl loflazepate Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(C(=O)OCC)N=C1C1=CC=CC=C1F CUCHJCMWNFEYOM-UHFFFAOYSA-N 0.000 description 1
- OGDVEMNWJVYAJL-LEPYJNQMSA-N Ethyl morphine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OCC OGDVEMNWJVYAJL-LEPYJNQMSA-N 0.000 description 1
- QQZWEECEMNQSTG-UHFFFAOYSA-N Ethyl nitrite Chemical compound CCON=O QQZWEECEMNQSTG-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- OGDVEMNWJVYAJL-UHFFFAOYSA-N Ethylmorphine Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OCC OGDVEMNWJVYAJL-UHFFFAOYSA-N 0.000 description 1
- DBVJJBKOTRCVKF-UHFFFAOYSA-N Etidronic acid Chemical compound OP(=O)(O)C(O)(C)P(O)(O)=O DBVJJBKOTRCVKF-UHFFFAOYSA-N 0.000 description 1
- HPHUVLMMVZITSG-UHFFFAOYSA-N Etiracetam Chemical compound CCC(C(N)=O)N1CCCC1=O HPHUVLMMVZITSG-UHFFFAOYSA-N 0.000 description 1
- VMZUTJCNQWMAGF-UHFFFAOYSA-N Etizolam Chemical compound S1C(CC)=CC2=C1N1C(C)=NN=C1CN=C2C1=CC=CC=C1Cl VMZUTJCNQWMAGF-UHFFFAOYSA-N 0.000 description 1
- 239000005896 Etofenprox Substances 0.000 description 1
- ITIONVBQFUNVJV-UHFFFAOYSA-N Etomidoline Chemical compound C12=CC=CC=C2C(=O)N(CC)C1NC(C=C1)=CC=C1OCCN1CCCCC1 ITIONVBQFUNVJV-UHFFFAOYSA-N 0.000 description 1
- KJTKYGFGPQSRRA-UHFFFAOYSA-N Etoxeridine Chemical compound C=1C=CC=CC=1C1(C(=O)OCC)CCN(CCOCCO)CC1 KJTKYGFGPQSRRA-UHFFFAOYSA-N 0.000 description 1
- WEEGYLXZBRQIMU-WAAGHKOSSA-N Eucalyptol Chemical compound C1C[C@H]2CC[C@]1(C)OC2(C)C WEEGYLXZBRQIMU-WAAGHKOSSA-N 0.000 description 1
- 239000005770 Eugenol Substances 0.000 description 1
- 101710089384 Extracellular protease Proteins 0.000 description 1
- 239000001116 FEMA 4028 Substances 0.000 description 1
- 108091008794 FGF receptors Proteins 0.000 description 1
- 108010079356 FIIa Proteins 0.000 description 1
- 108010023321 Factor VII Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- HMCCXLBXIJMERM-UHFFFAOYSA-N Febantel Chemical compound C1=C(NC(NC(=O)OC)=NC(=O)OC)C(NC(=O)COC)=CC(SC=2C=CC=CC=2)=C1 HMCCXLBXIJMERM-UHFFFAOYSA-N 0.000 description 1
- NIGWMJHCCYYCSF-UHFFFAOYSA-N Fenclonine Chemical compound OC(=O)C(N)CC1=CC=C(Cl)C=C1 NIGWMJHCCYYCSF-UHFFFAOYSA-N 0.000 description 1
- CLMFEXDZPBGYQQ-UHFFFAOYSA-N Fenestrel Chemical compound CCC1C(C)C(C(O)=O)CC=C1C1=CC=CC=C1 CLMFEXDZPBGYQQ-UHFFFAOYSA-N 0.000 description 1
- OVGORFFCBUIFIA-UHFFFAOYSA-N Fenipentol Chemical compound CCCCC(O)C1=CC=CC=C1 OVGORFFCBUIFIA-UHFFFAOYSA-N 0.000 description 1
- ASDCLYXOQCGHNT-UHFFFAOYSA-N Fenirofibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1C(O)C1=CC=C(Cl)C=C1 ASDCLYXOQCGHNT-UHFFFAOYSA-N 0.000 description 1
- RBBWCVQDXDFISW-UHFFFAOYSA-N Feprazone Chemical compound O=C1C(CC=C(C)C)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 RBBWCVQDXDFISW-UHFFFAOYSA-N 0.000 description 1
- IWQQNDAAVPMDJR-UOOQWULYSA-L Ferrotrenine Chemical compound O.O.[Fe++].C\C=N\C(C(C)O)C([O-])=O.C\C=N\C(C(C)O)C([O-])=O IWQQNDAAVPMDJR-UOOQWULYSA-L 0.000 description 1
- PMVSDNDAUGGCCE-TYYBGVCCSA-L Ferrous fumarate Chemical compound [Fe+2].[O-]C(=O)\C=C\C([O-])=O PMVSDNDAUGGCCE-TYYBGVCCSA-L 0.000 description 1
- SRANDCPSMJNFCK-WOJGMQOQSA-N Fezatione Chemical compound C1=CC(C)=CC=C1\C=N\N1C(=S)SC=C1C1=CC=CC=C1 SRANDCPSMJNFCK-WOJGMQOQSA-N 0.000 description 1
- 102000044168 Fibroblast Growth Factor Receptor Human genes 0.000 description 1
- 108090000368 Fibroblast growth factor 8 Proteins 0.000 description 1
- 229930183931 Filipin Natural products 0.000 description 1
- DJBNUMBKLMJRSA-UHFFFAOYSA-N Flecainide Chemical compound FC(F)(F)COC1=CC=C(OCC(F)(F)F)C(C(=O)NCC2NCCCC2)=C1 DJBNUMBKLMJRSA-UHFFFAOYSA-N 0.000 description 1
- APQPGQGAWABJLN-UHFFFAOYSA-N Floctafenine Chemical compound OCC(O)COC(=O)C1=CC=CC=C1NC1=CC=NC2=C(C(F)(F)F)C=CC=C12 APQPGQGAWABJLN-UHFFFAOYSA-N 0.000 description 1
- PTHLEKANMPKYDB-UHFFFAOYSA-N Flopropione Chemical compound CCC(=O)C1=C(O)C=C(O)C=C1O PTHLEKANMPKYDB-UHFFFAOYSA-N 0.000 description 1
- UIOFUWFRIANQPC-JKIFEVAISA-N Floxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(F)C=CC=C1Cl UIOFUWFRIANQPC-JKIFEVAISA-N 0.000 description 1
- CXLOIJUDIPVKOU-UHFFFAOYSA-N Fludorex Chemical compound CNCC(OC)C1=CC=CC(C(F)(F)F)=C1 CXLOIJUDIPVKOU-UHFFFAOYSA-N 0.000 description 1
- JBHUBOISLBWHAR-UHFFFAOYSA-N Flumezapine Chemical compound C1CN(C)CCN1C1=NC2=CC(F)=CC=C2NC2=C1C=C(C)S2 JBHUBOISLBWHAR-UHFFFAOYSA-N 0.000 description 1
- WJOHZNCJWYWUJD-IUGZLZTKSA-N Fluocinonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)COC(=O)C)[C@@]2(C)C[C@@H]1O WJOHZNCJWYWUJD-IUGZLZTKSA-N 0.000 description 1
- WHZRCUIISKRTJL-YTZKRAOUSA-N Fluocortolone caproate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)COC(=O)CCCCC)[C@@]2(C)C[C@@H]1O WHZRCUIISKRTJL-YTZKRAOUSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- OBWGMKKHCLHVIE-UHFFFAOYSA-N Fluperlapine Chemical compound C1CN(C)CCN1C1=NC2=CC(F)=CC=C2CC2=CC=CC=C12 OBWGMKKHCLHVIE-UHFFFAOYSA-N 0.000 description 1
- PLDUPXSUYLZYBN-UHFFFAOYSA-N Fluphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 PLDUPXSUYLZYBN-UHFFFAOYSA-N 0.000 description 1
- LRWSFOSWNAQHHW-UHFFFAOYSA-N Fluphenazine enanthate Chemical compound C1CN(CCOC(=O)CCCCCC)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 LRWSFOSWNAQHHW-UHFFFAOYSA-N 0.000 description 1
- WMFSSTNVXWNLKI-UHFFFAOYSA-N Flutazolam Chemical compound O1CCN2CC(=O)N(CCO)C3=CC=C(Cl)C=C3C21C1=CC=CC=C1F WMFSSTNVXWNLKI-UHFFFAOYSA-N 0.000 description 1
- RMFYWNFETXNTIQ-UHFFFAOYSA-N Flutemazepam Chemical compound N=1C(O)C(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F RMFYWNFETXNTIQ-UHFFFAOYSA-N 0.000 description 1
- OFVXPDXXVSGEPX-UHFFFAOYSA-N Flutoprazepam Chemical compound FC1=CC=CC=C1C(C1=CC(Cl)=CC=C11)=NCC(=O)N1CC1CC1 OFVXPDXXVSGEPX-UHFFFAOYSA-N 0.000 description 1
- 102000037909 Folate transporters Human genes 0.000 description 1
- 108091006783 Folate transporters Proteins 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- KGQZGCIVHYLPBH-UHFFFAOYSA-N Furafylline Chemical compound O=C1N(C)C(=O)C=2NC(C)=NC=2N1CC1=CC=CO1 KGQZGCIVHYLPBH-UHFFFAOYSA-N 0.000 description 1
- RGLLOUBXMOGLDQ-IVEVATEUSA-N Furazabol Chemical compound C([C@@H]1CC2)C3=NON=C3C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@](C)(O)[C@@]2(C)CC1 RGLLOUBXMOGLDQ-IVEVATEUSA-N 0.000 description 1
- IECPWNUMDGFDKC-UHFFFAOYSA-N Fusicsaeure Natural products C12C(O)CC3C(=C(CCC=C(C)C)C(O)=O)C(OC(C)=O)CC3(C)C1(C)CCC1C2(C)CCC(O)C1C IECPWNUMDGFDKC-UHFFFAOYSA-N 0.000 description 1
- 108091081406 G-quadruplex Proteins 0.000 description 1
- ZXRVKCBLGJOCEE-UHFFFAOYSA-N Gaboxadol Chemical compound C1NCCC2=C1ONC2=O ZXRVKCBLGJOCEE-UHFFFAOYSA-N 0.000 description 1
- XQLWNAFCTODIRK-UHFFFAOYSA-N Gallopamil Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC(OC)=C(OC)C(OC)=C1 XQLWNAFCTODIRK-UHFFFAOYSA-N 0.000 description 1
- ZPACYDRSPFRDHO-ROBAGEODSA-N Gefarnate Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\CCC(=O)OC\C=C(/C)CCC=C(C)C ZPACYDRSPFRDHO-ROBAGEODSA-N 0.000 description 1
- 229920001386 Gefarnate Polymers 0.000 description 1
- HEMJJKBWTPKOJG-UHFFFAOYSA-N Gemfibrozil Chemical compound CC1=CC=C(C)C(OCCCC(C)(C)C(O)=O)=C1 HEMJJKBWTPKOJG-UHFFFAOYSA-N 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- NMWQEPCLNXHPDX-UHFFFAOYSA-N Glybuzole Chemical compound S1C(C(C)(C)C)=NN=C1NS(=O)(=O)C1=CC=CC=C1 NMWQEPCLNXHPDX-UHFFFAOYSA-N 0.000 description 1
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 1
- 239000008777 Glycerylphosphorylcholine Substances 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- HNSCCNJWTJUGNQ-UHFFFAOYSA-N Glyclopyramide Chemical compound C1=CC(Cl)=CC=C1S(=O)(=O)NC(=O)NN1CCCC1 HNSCCNJWTJUGNQ-UHFFFAOYSA-N 0.000 description 1
- 108010015899 Glycopeptides Proteins 0.000 description 1
- 102000002068 Glycopeptides Human genes 0.000 description 1
- VPNYRYCIDCJBOM-UHFFFAOYSA-M Glycopyrronium bromide Chemical compound [Br-].C1[N+](C)(C)CCC1OC(=O)C(O)(C=1C=CC=CC=1)C1CCCC1 VPNYRYCIDCJBOM-UHFFFAOYSA-M 0.000 description 1
- 229920002683 Glycosaminoglycan Polymers 0.000 description 1
- MPDGHEJMBKOTSU-UHFFFAOYSA-N Glycyrrhetinsaeure Natural products C12C(=O)C=C3C4CC(C)(C(O)=O)CCC4(C)CCC3(C)C1(C)CCC1C2(C)CCC(O)C1(C)C MPDGHEJMBKOTSU-UHFFFAOYSA-N 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 102000016355 Granulocyte-Macrophage Colony-Stimulating Factor Receptors Human genes 0.000 description 1
- 108010092372 Granulocyte-Macrophage Colony-Stimulating Factor Receptors Proteins 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- HSRJKNPTNIJEKV-UHFFFAOYSA-N Guaifenesin Chemical compound COC1=CC=CC=C1OCC(O)CO HSRJKNPTNIJEKV-UHFFFAOYSA-N 0.000 description 1
- WDZVGELJXXEGPV-YIXHJXPBSA-N Guanabenz Chemical compound NC(N)=N\N=C\C1=C(Cl)C=CC=C1Cl WDZVGELJXXEGPV-YIXHJXPBSA-N 0.000 description 1
- INJOMKTZOLKMBF-UHFFFAOYSA-N Guanfacine Chemical compound NC(=N)NC(=O)CC1=C(Cl)C=CC=C1Cl INJOMKTZOLKMBF-UHFFFAOYSA-N 0.000 description 1
- QXZGBUJJYSLZLT-UHFFFAOYSA-N H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH Natural products NC(N)=NCCCC(N)C(=O)N1CCCC1C(=O)N1C(C(=O)NCC(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CO)C(=O)N2C(CCC2)C(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CCCN=C(N)N)C(O)=O)CCC1 QXZGBUJJYSLZLT-UHFFFAOYSA-N 0.000 description 1
- WYCLKVQLVUQKNZ-UHFFFAOYSA-N Halazepam Chemical compound N=1CC(=O)N(CC(F)(F)F)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 WYCLKVQLVUQKNZ-UHFFFAOYSA-N 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 101710121697 Heat-stable enterotoxin Proteins 0.000 description 1
- 229920001499 Heparinoid Polymers 0.000 description 1
- GVGLGOZIDCSQPN-PVHGPHFFSA-N Heroin Chemical compound O([C@H]1[C@H](C=C[C@H]23)OC(C)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4OC(C)=O GVGLGOZIDCSQPN-PVHGPHFFSA-N 0.000 description 1
- 108010007267 Hirudins Proteins 0.000 description 1
- 102000007625 Hirudins Human genes 0.000 description 1
- 101000945318 Homo sapiens Calponin-1 Proteins 0.000 description 1
- 101000595923 Homo sapiens Placenta growth factor Proteins 0.000 description 1
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 229920001612 Hydroxyethyl starch Polymers 0.000 description 1
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 1
- 108010031794 IGF Type 1 Receptor Proteins 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000005795 Imazalil Substances 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 1
- 102100025305 Integrin alpha-2 Human genes 0.000 description 1
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 1
- 108010064600 Intercellular Adhesion Molecule-3 Proteins 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 102100037872 Intercellular adhesion molecule 2 Human genes 0.000 description 1
- 101710148794 Intercellular adhesion molecule 2 Proteins 0.000 description 1
- 102100037871 Intercellular adhesion molecule 3 Human genes 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 1
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 1
- 108010065805 Interleukin-12 Proteins 0.000 description 1
- 102000013462 Interleukin-12 Human genes 0.000 description 1
- 102000004125 Interleukin-1alpha Human genes 0.000 description 1
- 108010082786 Interleukin-1alpha Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 1
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 102000004890 Interleukin-8 Human genes 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 229920001202 Inulin Polymers 0.000 description 1
- UXIGWFXRQKWHHA-UHFFFAOYSA-N Iotalamic acid Chemical compound CNC(=O)C1=C(I)C(NC(C)=O)=C(I)C(C(O)=O)=C1I UXIGWFXRQKWHHA-UHFFFAOYSA-N 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- KQEOKUJOWVGWDX-UHFFFAOYSA-N Isodihydrovaltrate Natural products CC(C)CC(=O)OC1CC(C(=COC2OC(=O)CC(C)C)COC(C)=O)C2C11CO1 KQEOKUJOWVGWDX-UHFFFAOYSA-N 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- 108010092694 L-Selectin Proteins 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- LEVWYRKDKASIDU-IMJSIDKUSA-N L-cystine Chemical compound [O-]C(=O)[C@@H]([NH3+])CSSC[C@H]([NH3+])C([O-])=O LEVWYRKDKASIDU-IMJSIDKUSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 102100033467 L-selectin Human genes 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- ZZJYIKPMDIWRSN-HWBMXIPRSA-N LSM-20934 Chemical compound C12=CC=CC=C2CCC2=CC=CC3=C2[C@H]1CN1CC[C@](C(C)(C)C)(O)C[C@H]13 ZZJYIKPMDIWRSN-HWBMXIPRSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 108010000851 Laminin Receptors Proteins 0.000 description 1
- 102000002297 Laminin Receptors Human genes 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- HLFSDGLLUJUHTE-SNVBAGLBSA-N Levamisole Chemical compound C1([C@H]2CN3CCSC3=N2)=CC=CC=C1 HLFSDGLLUJUHTE-SNVBAGLBSA-N 0.000 description 1
- ZCGOMHNNNFPNMX-YHYDXASRSA-N Levocabastinum Chemical compound C1([C@@]2(C(O)=O)CCN(C[C@H]2C)C2CCC(CC2)(C#N)C=2C=CC(F)=CC=2)=CC=CC=C1 ZCGOMHNNNFPNMX-YHYDXASRSA-N 0.000 description 1
- ODLGFPIWRAEFAN-PFEQFJNWSA-N Levomepromazine hydrochloride Chemical compound Cl.C1=CC=C2N(C[C@H](C)CN(C)C)C3=CC(OC)=CC=C3SC2=C1 ODLGFPIWRAEFAN-PFEQFJNWSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- OJMMVQQUTAEWLP-UHFFFAOYSA-N Lincomycin Natural products CN1CC(CCC)CC1C(=O)NC(C(C)O)C1C(O)C(O)C(O)C(SC)O1 OJMMVQQUTAEWLP-UHFFFAOYSA-N 0.000 description 1
- 102000004882 Lipase Human genes 0.000 description 1
- 108090001060 Lipase Proteins 0.000 description 1
- 239000004367 Lipase Substances 0.000 description 1
- 239000000232 Lipid Bilayer Substances 0.000 description 1
- WVVSZNPYNCNODU-XTQGRXLLSA-N Lysergic acid propanolamide Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@H](CO)C)C2)=C3C2=CNC3=C1 WVVSZNPYNCNODU-XTQGRXLLSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 101710145006 Lysis protein Proteins 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100028123 Macrophage colony-stimulating factor 1 Human genes 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 1
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 1
- 101800001751 Melanocyte-stimulating hormone alpha Proteins 0.000 description 1
- 102100027159 Membrane primary amine oxidase Human genes 0.000 description 1
- 101710132836 Membrane primary amine oxidase Proteins 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- SIDLZWOQUZRBRU-UHFFFAOYSA-N Methyprylon Chemical compound CCC1(CC)C(=O)NCC(C)C1=O SIDLZWOQUZRBRU-UHFFFAOYSA-N 0.000 description 1
- DIGFQJFCDPKEPF-OIUSMDOTSA-L Metocurine iodide Chemical compound [I-].[I-].C1([C@@H]([N+](CCC1=CC=1OC)(C)C)CC2=CC=C(C=C2)O2)=CC=1OC(=C1)C(OC)=CC=C1C[C@H]1[N+](C)(C)CCC3=C1C2=C(OC)C(OC)=C3 DIGFQJFCDPKEPF-OIUSMDOTSA-L 0.000 description 1
- BAQCROVBDNBEEB-UBYUBLNFSA-N Metrizamide Chemical compound CC(=O)N(C)C1=C(I)C(NC(C)=O)=C(I)C(C(=O)N[C@@H]2[C@H]([C@H](O)[C@@H](CO)OC2O)O)=C1I BAQCROVBDNBEEB-UBYUBLNFSA-N 0.000 description 1
- BYBLEWFAAKGYCD-UHFFFAOYSA-N Miconazole Chemical compound ClC1=CC(Cl)=CC=C1COC(C=1C(=CC(Cl)=CC=1)Cl)CN1C=NC=C1 BYBLEWFAAKGYCD-UHFFFAOYSA-N 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 102000005431 Molecular Chaperones Human genes 0.000 description 1
- 108010006519 Molecular Chaperones Proteins 0.000 description 1
- 101710167839 Morphogenetic protein Proteins 0.000 description 1
- 102000015728 Mucins Human genes 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 102000005717 Myeloma Proteins Human genes 0.000 description 1
- 108010045503 Myeloma Proteins Proteins 0.000 description 1
- 102000003505 Myosin Human genes 0.000 description 1
- 108060008487 Myosin Proteins 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- ICKFFNBDFNZJSX-UHFFFAOYSA-N N'-[(4-chlorophenyl)methyl]-N,N-dimethyl-N'-(2-pyridinyl)ethane-1,2-diamine Chemical compound C=1C=CC=NC=1N(CCN(C)C)CC1=CC=C(Cl)C=C1 ICKFFNBDFNZJSX-UHFFFAOYSA-N 0.000 description 1
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 1
- MMOXZBCLCQITDF-UHFFFAOYSA-N N,N-diethyl-m-toluamide Chemical compound CCN(CC)C(=O)C1=CC=CC(C)=C1 MMOXZBCLCQITDF-UHFFFAOYSA-N 0.000 description 1
- JTAJFHGSVCEPKC-KUHUBIRLSA-N N,N-dimethyl-3-[(9S,10R)-10-methyl-2-(trifluoromethyl)-9,10-dihydroanthracen-9-yl]propan-1-amine Chemical compound FC(F)(F)C1=CC=C2[C@H](C)C3=CC=CC=C3[C@H](CCCN(C)C)C2=C1 JTAJFHGSVCEPKC-KUHUBIRLSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical class ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- JUUFBMODXQKSTD-UHFFFAOYSA-N N-[2-amino-6-[(4-fluorophenyl)methylamino]-3-pyridinyl]carbamic acid ethyl ester Chemical compound N1=C(N)C(NC(=O)OCC)=CC=C1NCC1=CC=C(F)C=C1 JUUFBMODXQKSTD-UHFFFAOYSA-N 0.000 description 1
- 125000005118 N-alkylcarbamoyl group Chemical group 0.000 description 1
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 description 1
- UIAYVIIHMORPSJ-UHFFFAOYSA-N N-cyclohexyl-N-methyl-4-[(2-oxo-1H-quinolin-6-yl)oxy]butanamide Chemical compound C=1C=C2NC(=O)C=CC2=CC=1OCCCC(=O)N(C)C1CCCCC1 UIAYVIIHMORPSJ-UHFFFAOYSA-N 0.000 description 1
- QWZFVMCWPLMLTL-UHFFFAOYSA-N N-diaminophosphoryl-4-fluorobenzamide Chemical compound NP(N)(=O)NC(=O)C1=CC=C(F)C=C1 QWZFVMCWPLMLTL-UHFFFAOYSA-N 0.000 description 1
- GHAZCVNUKKZTLG-UHFFFAOYSA-N N-ethyl-succinimide Natural products CCN1C(=O)CCC1=O GHAZCVNUKKZTLG-UHFFFAOYSA-N 0.000 description 1
- HDFGOPSGAURCEO-UHFFFAOYSA-N N-ethylmaleimide Chemical compound CCN1C(=O)C=CC1=O HDFGOPSGAURCEO-UHFFFAOYSA-N 0.000 description 1
- QSQQPMHPCBLLGX-UHFFFAOYSA-N N-methyl-4-[2-(phenylmethyl)phenoxy]-1-butanamine Chemical compound CNCCCCOC1=CC=CC=C1CC1=CC=CC=C1 QSQQPMHPCBLLGX-UHFFFAOYSA-N 0.000 description 1
- YUGZHQHSNYIFLG-UHFFFAOYSA-N N-phenylcarbamic acid [2-[anilino(oxo)methoxy]-3-(1-piperidinyl)propyl] ester Chemical compound C1CCCCN1CC(OC(=O)NC=1C=CC=CC=1)COC(=O)NC1=CC=CC=C1 YUGZHQHSNYIFLG-UHFFFAOYSA-N 0.000 description 1
- DRBBFCLWYRJSJZ-UHFFFAOYSA-N N-phosphocreatine Chemical compound OC(=O)CN(C)C(=N)NP(O)(O)=O DRBBFCLWYRJSJZ-UHFFFAOYSA-N 0.000 description 1
- JMPFSEBWVLAJKM-UHFFFAOYSA-N N-{5-chloro-4-[(4-chlorophenyl)(cyano)methyl]-2-methylphenyl}-2-hydroxy-3,5-diiodobenzamide Chemical compound ClC=1C=C(NC(=O)C=2C(=C(I)C=C(I)C=2)O)C(C)=CC=1C(C#N)C1=CC=C(Cl)C=C1 JMPFSEBWVLAJKM-UHFFFAOYSA-N 0.000 description 1
- 108091008604 NGF receptors Proteins 0.000 description 1
- IRQXZTBHNKVIRL-UHFFFAOYSA-N NSC 165563 Natural products CC1=C(O)C(=O)CC2(C)C(C(O)C3O)C45COC3(C(=O)OC)C5C(OC(=O)C=C(C)C(C)C)C(=O)OC4CC21 IRQXZTBHNKVIRL-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 102000007339 Nerve Growth Factor Receptors Human genes 0.000 description 1
- 108010042309 Netropsin Proteins 0.000 description 1
- 102000005348 Neuraminidase Human genes 0.000 description 1
- 108010006232 Neuraminidase Proteins 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- SNIOPGDIGTZGOP-UHFFFAOYSA-N Nitroglycerin Chemical compound [O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=O SNIOPGDIGTZGOP-UHFFFAOYSA-N 0.000 description 1
- 239000000006 Nitroglycerin Substances 0.000 description 1
- 108010070047 Notch Receptors Proteins 0.000 description 1
- BXEFQPCKQSTMKA-UHFFFAOYSA-N OC(=O)C=[N+]=[N-] Chemical class OC(=O)C=[N+]=[N-] BXEFQPCKQSTMKA-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 239000008896 Opium Substances 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 102000004264 Osteopontin Human genes 0.000 description 1
- 108010081689 Osteopontin Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- LCAHPIFLPICNRW-SVYNMNNPSA-N Oxymetebanol Chemical compound C1[C@H](O)CC[C@@]2(O)[C@H]3CC4=CC=C(OC)C(OC)=C4[C@]21CCN3C LCAHPIFLPICNRW-SVYNMNNPSA-N 0.000 description 1
- 102100028139 Oxytocin receptor Human genes 0.000 description 1
- 108090000876 Oxytocin receptors Proteins 0.000 description 1
- 108010035766 P-Selectin Proteins 0.000 description 1
- 102100023472 P-selectin Human genes 0.000 description 1
- 102100037600 P2Y purinoceptor 1 Human genes 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 241000282320 Panthera leo Species 0.000 description 1
- HYRKAAMZBDSJFJ-LFDBJOOHSA-N Paramethasone acetate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)COC(C)=O)(O)[C@@]2(C)C[C@@H]1O HYRKAAMZBDSJFJ-LFDBJOOHSA-N 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- TZRXHJWUDPFEEY-UHFFFAOYSA-N Pentaerythritol Tetranitrate Chemical compound [O-][N+](=O)OCC(CO[N+]([O-])=O)(CO[N+]([O-])=O)CO[N+]([O-])=O TZRXHJWUDPFEEY-UHFFFAOYSA-N 0.000 description 1
- 239000000026 Pentaerythritol tetranitrate Substances 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- AJOQSQHYDOFIOX-UHFFFAOYSA-N Pheneturide Chemical compound NC(=O)NC(=O)C(CC)C1=CC=CC=C1 AJOQSQHYDOFIOX-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 102100035194 Placenta growth factor Human genes 0.000 description 1
- 102100024616 Platelet endothelial cell adhesion molecule Human genes 0.000 description 1
- 229920001744 Polyaldehyde Polymers 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 108010040201 Polymyxins Proteins 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000007324 Protease Nexins Human genes 0.000 description 1
- 108010007544 Protease Nexins Proteins 0.000 description 1
- UVMRYBDEERADNV-UHFFFAOYSA-N Pseudoeugenol Natural products COC1=CC(C(C)=C)=CC=C1O UVMRYBDEERADNV-UHFFFAOYSA-N 0.000 description 1
- 108010085249 Purinergic P2 Receptors Proteins 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 101710132082 Pyrimidine/purine nucleoside phosphorylase Proteins 0.000 description 1
- 102000044126 RNA-Binding Proteins Human genes 0.000 description 1
- 108700020471 RNA-Binding Proteins Proteins 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- PPTYJKAXVCCBDU-UHFFFAOYSA-N Rohypnol Chemical compound N=1CC(=O)N(C)C2=CC=C([N+]([O-])=O)C=C2C=1C1=CC=CC=C1F PPTYJKAXVCCBDU-UHFFFAOYSA-N 0.000 description 1
- GBFLZEXEOZUWRN-VKHMYHEASA-N S-carboxymethyl-L-cysteine Chemical compound OC(=O)[C@@H](N)CSCC(O)=O GBFLZEXEOZUWRN-VKHMYHEASA-N 0.000 description 1
- AUVVAXYIELKVAI-UHFFFAOYSA-N SJ000285215 Natural products N1CCC2=CC(OC)=C(OC)C=C2C1CC1CC2C3=CC(OC)=C(OC)C=C3CCN2CC1CC AUVVAXYIELKVAI-UHFFFAOYSA-N 0.000 description 1
- ZRIHAIZYIMGOAB-UHFFFAOYSA-N Secbutobarbitone Natural products CCC(C)C1(CC)C(=O)NC(=O)NC1=O ZRIHAIZYIMGOAB-UHFFFAOYSA-N 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 101710104074 Spermidine/putrescine-binding periplasmic protein Proteins 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- CPFNIKYEDJFRAT-UHFFFAOYSA-N Strospasid Natural products OC1C(OC)C(O)C(C)OC1OC1CC(CCC2C3(CC(O)C(C3(C)CCC32)C=2COC(=O)C=2)O)C3(C)CC1 CPFNIKYEDJFRAT-UHFFFAOYSA-N 0.000 description 1
- 244000028419 Styrax benzoin Species 0.000 description 1
- 235000000126 Styrax benzoin Nutrition 0.000 description 1
- 102400000096 Substance P Human genes 0.000 description 1
- 101800003906 Substance P Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical group O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 1
- 235000008411 Sumatra benzointree Nutrition 0.000 description 1
- 102000019361 Syndecan Human genes 0.000 description 1
- 108050006774 Syndecan Proteins 0.000 description 1
- 229920001963 Synthetic biodegradable polymer Polymers 0.000 description 1
- CYQFCXCEBYINGO-UHFFFAOYSA-N THC Natural products C1=C(C)CCC2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3C21 CYQFCXCEBYINGO-UHFFFAOYSA-N 0.000 description 1
- 108010006830 TIE receptors Proteins 0.000 description 1
- 102000005450 TIE receptors Human genes 0.000 description 1
- SEQDDYPDSLOBDC-UHFFFAOYSA-N Temazepam Chemical compound N=1C(O)C(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 SEQDDYPDSLOBDC-UHFFFAOYSA-N 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- NSOXQYCFHDMMGV-UHFFFAOYSA-N Tetrakis(2-hydroxypropyl)ethylenediamine Chemical compound CC(O)CN(CC(C)O)CCN(CC(C)O)CC(C)O NSOXQYCFHDMMGV-UHFFFAOYSA-N 0.000 description 1
- 241000906446 Theraps Species 0.000 description 1
- IUJDSEJGGMCXSG-UHFFFAOYSA-N Thiopental Chemical compound CCCC(C)C1(CC)C(=O)NC(=S)NC1=O IUJDSEJGGMCXSG-UHFFFAOYSA-N 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical group OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 108090000166 Thrombin receptors Proteins 0.000 description 1
- 102000003790 Thrombin receptors Human genes 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 102100031372 Thymidine phosphorylase Human genes 0.000 description 1
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 1
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 1
- 108050006955 Tissue-type plasminogen activator Proteins 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 102100033142 Transcription factor 20 Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 108060008539 Transglutaminase Proteins 0.000 description 1
- UMILHIMHKXVDGH-UHFFFAOYSA-N Triethylene glycol diglycidyl ether Chemical compound C1OC1COCCOCCOCCOCC1CO1 UMILHIMHKXVDGH-UHFFFAOYSA-N 0.000 description 1
- GPLGAQQQNWMVMM-UHFFFAOYSA-N Trimethyl-dihydro-conkurchin Natural products C1C=C2CC(N(C)C)CCC2(C)C2C1C1CCC3C(C)N(C)CC31CC2 GPLGAQQQNWMVMM-UHFFFAOYSA-N 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 1
- 206010054094 Tumour necrosis Diseases 0.000 description 1
- CKNOLMVLQUPVMU-UHFFFAOYSA-N UNPD183315 Natural products O1C(C)C(OC2C(C(O)C(O)C(CO)O2)O)C(OC)C(O)C1OC(C1)CCC2(C)C1CCC(C1(CC3O)O)C2CCC1(C)C3C1=CC(=O)OC1 CKNOLMVLQUPVMU-UHFFFAOYSA-N 0.000 description 1
- VGQOVCHZGQWAOI-UHFFFAOYSA-N UNPD55612 Natural products N1C(O)C2CC(C=CC(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-UHFFFAOYSA-N 0.000 description 1
- 108010053096 Vascular Endothelial Growth Factor Receptor-1 Proteins 0.000 description 1
- 102000016663 Vascular Endothelial Growth Factor Receptor-3 Human genes 0.000 description 1
- 108010053100 Vascular Endothelial Growth Factor Receptor-3 Proteins 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 108010003205 Vasoactive Intestinal Peptide Proteins 0.000 description 1
- 102400000015 Vasoactive intestinal peptide Human genes 0.000 description 1
- 102000004136 Vasopressin Receptors Human genes 0.000 description 1
- 108090000643 Vasopressin Receptors Proteins 0.000 description 1
- 108010004977 Vasopressins Proteins 0.000 description 1
- 102000002852 Vasopressins Human genes 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- MECHNRXZTMCUDQ-UHFFFAOYSA-N Vitamin D2 Natural products C1CCC2(C)C(C(C)C=CC(C)C(C)C)CCC2C1=CC=C1CC(O)CCC1=C MECHNRXZTMCUDQ-UHFFFAOYSA-N 0.000 description 1
- 102100023038 WD and tetratricopeptide repeats protein 1 Human genes 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 102100033220 Xanthine oxidase Human genes 0.000 description 1
- 108010093894 Xanthine oxidase Proteins 0.000 description 1
- WREGKURFCTUGRC-POYBYMJQSA-N Zalcitabine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)CC1 WREGKURFCTUGRC-POYBYMJQSA-N 0.000 description 1
- ZMQRJWIYMXZORG-GZIFKOAOSA-N [(1e,3r,4r,6r,7z,9z,11e)-3,6,13-trihydroxy-3-methyl-1-[(2s)-6-oxo-2,3-dihydropyran-2-yl]trideca-1,7,9,11-tetraen-4-yl] dihydrogen phosphate Chemical compound OC/C=C/C=C\C=C/[C@H](O)C[C@@H](OP(O)(O)=O)[C@@](O)(C)\C=C\[C@@H]1CC=CC(=O)O1 ZMQRJWIYMXZORG-GZIFKOAOSA-N 0.000 description 1
- IYBYNRHXGXDDDS-VRRJBYJJSA-N [(1r,4as,4bs,10ar,10bs,11s,12as)-1-acetyl-11-hydroxy-10a,12a-dimethyl-8-oxo-2,3,4,4a,4b,5,6,10b,11,12-decahydrochrysen-1-yl] butanoate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CCC[C@@](C(C)=O)(OC(=O)CCC)[C@@]1(C)C[C@@H]2O IYBYNRHXGXDDDS-VRRJBYJJSA-N 0.000 description 1
- JABDOYKGZCPHPX-LVJITLAUSA-M [(1r,5s)-8-[(4-butoxyphenyl)methyl]-8-methyl-8-azoniabicyclo[3.2.1]octan-3-yl] (2s)-3-hydroxy-2-phenylpropanoate;bromide Chemical compound [Br-].C1=CC(OCCCC)=CC=C1C[N+]1(C)[C@@H]2CC[C@H]1CC(OC(=O)[C@H](CO)C=1C=CC=CC=1)C2 JABDOYKGZCPHPX-LVJITLAUSA-M 0.000 description 1
- RQLDKUSQKQMFCN-DOOXDOMHSA-N [(1s,2s,3s,4s,5r,6r)-3-(diaminomethylideneamino)-4-[(2r,3r,4r,5s)-3-[(2s,3s,4s,5r,6s)-4,5-dihydroxy-6-(hydroxymethyl)-3-(methylamino)oxan-2-yl]oxy-4-hydroxy-4-(hydroxymethyl)-5-methyloxolan-2-yl]oxy-2,5,6-trihydroxycyclohexyl] carbamate Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](CO)(O)[C@H](C)O[C@H]1O[C@H]1[C@@H](N=C(N)N)[C@H](O)[C@H](OC(N)=O)[C@H](O)[C@H]1O RQLDKUSQKQMFCN-DOOXDOMHSA-N 0.000 description 1
- JSZILQVIPPROJI-CEXWTWQISA-N [(2R,3R,11bS)-3-(diethylcarbamoyl)-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-benzo[a]quinolizin-2-yl] acetate Chemical compound C1CC2=CC(OC)=C(OC)C=C2[C@H]2N1C[C@@H](C(=O)N(CC)CC)[C@H](OC(C)=O)C2 JSZILQVIPPROJI-CEXWTWQISA-N 0.000 description 1
- QNYRJYLGBCLNFA-QFIPXVFZSA-N [(2r)-3-[(dimethylamino)methyl]-1,2-diphenylbut-3-en-2-yl] propanoate Chemical compound C([C@](OC(=O)CC)(C(=C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 QNYRJYLGBCLNFA-QFIPXVFZSA-N 0.000 description 1
- UFUVLHLTWXBHGZ-MGZQPHGTSA-N [(2r,3r,4s,5r,6r)-6-[(1s,2s)-2-chloro-1-[[(2s,4r)-1-methyl-4-propylpyrrolidine-2-carbonyl]amino]propyl]-4,5-dihydroxy-2-methylsulfanyloxan-3-yl] dihydrogen phosphate Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@H](C)Cl)[C@@H]1[C@H](O)[C@H](O)[C@@H](OP(O)(O)=O)[C@@H](SC)O1 UFUVLHLTWXBHGZ-MGZQPHGTSA-N 0.000 description 1
- SPKNARKFCOPTSY-XWPZMVOTSA-N [(2r,3s)-2-[(2s,3r)-3-methyloxiran-2-yl]-6-oxo-2,3-dihydropyran-3-yl] acetate Chemical compound C[C@H]1O[C@@H]1[C@H]1[C@@H](OC(C)=O)C=CC(=O)O1 SPKNARKFCOPTSY-XWPZMVOTSA-N 0.000 description 1
- ZLNMZWDQJJAEAM-QPBDZWBWSA-N [(2r,3s,4r,5r,6s)-5-(pyridine-3-carbonylamino)-3,4,6-tris(pyridine-3-carbonyloxy)oxan-2-yl]methyl pyridine-3-carboxylate Chemical compound O([C@H]1[C@@H]([C@H]([C@H](OC(=O)C=2C=NC=CC=2)[C@@H](COC(=O)C=2C=NC=CC=2)O1)OC(=O)C=1C=NC=CC=1)NC(=O)C=1C=NC=CC=1)C(=O)C1=CC=CN=C1 ZLNMZWDQJJAEAM-QPBDZWBWSA-N 0.000 description 1
- FPEVNWDVXZGKCD-SAVXWCSHSA-N [(2s,3r,4s,6r)-4-(dimethylamino)-2-[[(3r,4s,5s,6r,7r,9r,11r,12r,13s,14r)-14-ethyl-7,12,13-trihydroxy-4-[(2r,4r,5s,6s)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy-3,5,7,9,11,13-hexamethyl-2,10-dioxo-oxacyclotetradec-6-yl]oxy]-6-methyloxan-3-yl] acetate;o Chemical compound CCCCCCCCCCCCCCCCCC(O)=O.O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)OC(C)=O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 FPEVNWDVXZGKCD-SAVXWCSHSA-N 0.000 description 1
- FQVHOULQCKDUCY-OGHXVOSASA-N [(2s,3s,4r,6s)-6-[(2r,3s,4r,5r,6s)-6-[[(1s,3r,7r,8s,9s,10r,12r,14e,16s)-7-acetyloxy-8-methoxy-3,12-dimethyl-5,13-dioxo-10-(2-oxoethyl)-4,17-dioxabicyclo[14.1.0]heptadec-14-en-9-yl]oxy]-4-(dimethylamino)-5-hydroxy-2-methyloxan-3-yl]oxy-4-hydroxy-2,4-dimeth Chemical compound O([C@@H]1[C@@H](C)O[C@H]([C@@H]([C@H]1N(C)C)O)O[C@H]1[C@@H](CC=O)C[C@@H](C)C(=O)/C=C/[C@@H]2O[C@H]2C[C@@H](C)OC(=O)C[C@H]([C@@H]1OC)OC(C)=O)[C@H]1C[C@@](C)(O)[C@@H](OC(=O)CC(C)C)[C@H](C)O1 FQVHOULQCKDUCY-OGHXVOSASA-N 0.000 description 1
- SUKULMJPMHYWKC-GMMLHHOXSA-L [(2s,3s,5s,8r,9s,10s,13s,14s,16s,17r)-17-hydroxy-10,13-dimethyl-2,16-bis(1-methylpiperidin-1-ium-1-yl)-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl] acetate;dibromide Chemical compound [Br-].[Br-].C[N+]1([C@H]2C[C@]3(C)[C@H]4CC[C@]5(C)[C@@H](O)[C@H](C[C@H]5[C@@H]4CC[C@H]3C[C@@H]2OC(=O)C)[N+]2(C)CCCCC2)CCCCC1 SUKULMJPMHYWKC-GMMLHHOXSA-L 0.000 description 1
- YASNUXZKZNVXIS-CBNXCZCTSA-N [(3ar,6r,6ar)-4-[[2-chloroethyl(nitroso)carbamoyl]amino]-2,2-dimethyl-3a,4,6,6a-tetrahydrofuro[3,4-d][1,3]dioxol-6-yl]methyl 4-nitrobenzoate Chemical compound C([C@@H]1[C@H]2OC(O[C@H]2C(NC(=O)N(CCCl)N=O)O1)(C)C)OC(=O)C1=CC=C([N+]([O-])=O)C=C1 YASNUXZKZNVXIS-CBNXCZCTSA-N 0.000 description 1
- MEFAKOINNJLWFO-MAZIBIHTSA-N [(3r)-4-[[3-[2-[2-[3-[[(2s)-3,3-dimethyl-2,4-bis(pyridine-3-carbonyloxy)butanoyl]amino]propanoylamino]ethyldisulfanyl]ethylamino]-3-oxopropyl]amino]-2,2-dimethyl-4-oxo-3-(pyridine-3-carbonyloxy)butyl] pyridine-3-carboxylate Chemical compound CC(C)([C@@H](OC(=O)C=1C=NC=CC=1)C(=O)NCCC(=O)NCCSSCCNC(=O)CCNC(=O)[C@@H](OC(=O)C=1C=NC=CC=1)C(C)(C)COC(=O)C=1C=NC=CC=1)COC(=O)C1=CC=CN=C1 MEFAKOINNJLWFO-MAZIBIHTSA-N 0.000 description 1
- ODEGQXRCQDVXSJ-RHSMWYFYSA-N [(3r,4r)-3-ethyl-1-methyl-4-phenylpiperidin-4-yl] propanoate Chemical compound CC[C@@H]1CN(C)CC[C@]1(OC(=O)CC)C1=CC=CC=C1 ODEGQXRCQDVXSJ-RHSMWYFYSA-N 0.000 description 1
- WTIJXIZOODAMJT-WBACWINTSA-N [(3r,4s,5r,6s)-5-hydroxy-6-[4-hydroxy-3-[[5-[[4-hydroxy-7-[(2s,3r,4s,5r)-3-hydroxy-5-methoxy-6,6-dimethyl-4-(5-methyl-1h-pyrrole-2-carbonyl)oxyoxan-2-yl]oxy-8-methyl-2-oxochromen-3-yl]carbamoyl]-4-methyl-1h-pyrrole-3-carbonyl]amino]-8-methyl-2-oxochromen- Chemical compound O([C@@H]1[C@H](C(O[C@H](OC=2C(=C3OC(=O)C(NC(=O)C=4C(=C(C(=O)NC=5C(OC6=C(C)C(O[C@@H]7[C@@H]([C@H](OC(=O)C=8NC(C)=CC=8)[C@@H](OC)C(C)(C)O7)O)=CC=C6C=5O)=O)NC=4)C)=C(O)C3=CC=2)C)[C@@H]1O)(C)C)OC)C(=O)C1=CC=C(C)N1 WTIJXIZOODAMJT-WBACWINTSA-N 0.000 description 1
- HYQJLIGADHPYIR-MVEXVFSBSA-N [(3r,5s,6s,7r,8s,9s,12r,13s,14s,15r)-6-[(2s,3r,4s,6r)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-8-[(2r,4s,5s,6s)-4-methoxy-6-methyl-5-propanoyloxyoxan-2-yl]oxy-5,7,9,12,13,15-hexamethyl-10,16-dioxo-1,11-dioxaspiro[2.13]hexadecan-14-yl] propanoate Chemical compound C1[C@H](OC)[C@@H](OC(=O)CC)[C@H](C)O[C@H]1O[C@@H]1[C@H](C)C(=O)O[C@H](C)[C@H](C)[C@H](OC(=O)CC)[C@@H](C)C(=O)[C@@]2(OC2)C[C@H](C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C HYQJLIGADHPYIR-MVEXVFSBSA-N 0.000 description 1
- UVAZQQHAVMNMHE-BBRMVZONSA-N [(3s,4s)-1,3-dimethyl-4-phenylpiperidin-4-yl] propanoate Chemical compound C=1C=CC=CC=1[C@]1(OC(=O)CC)CCN(C)C[C@@H]1C UVAZQQHAVMNMHE-BBRMVZONSA-N 0.000 description 1
- KSCZWFXQKITHSL-OKCNGXCSSA-N [(3s,8r,9s,10r,13s,14s,17r)-17-acetyl-17-acetyloxy-6-chloro-10,13-dimethyl-1,2,3,8,9,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-3-yl] acetate Chemical compound C1C[C@]2(C)[C@](C(C)=O)(OC(C)=O)CC[C@H]2[C@@H]2C=C(Cl)C3=C[C@@H](OC(=O)C)CC[C@]3(C)[C@H]21 KSCZWFXQKITHSL-OKCNGXCSSA-N 0.000 description 1
- PNEOEUCBHVHXHF-NFQWVWSDSA-N [(5R,8S,9S,10R,13S,14S)-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-4-yl] propanoate Chemical compound C(CC)(=O)OC1[C@@H]2CC[C@@H]3[C@H](CC[C@@]4(CCC[C@H]43)C)[C@]2(CCC1)C PNEOEUCBHVHXHF-NFQWVWSDSA-N 0.000 description 1
- MMPCMJGFURZYOY-WRWLIDTKSA-N [(6ar,9r,10ar)-5-bromo-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline-9-yl]methyl azepane-1-carboxylate Chemical compound C([C@H]1CN([C@H]2[C@@H](C=3C=CC=C4NC(Br)=C(C=34)C2)C1)C)OC(=O)N1CCCCCC1 MMPCMJGFURZYOY-WRWLIDTKSA-N 0.000 description 1
- MVLBCBPGBUAVJQ-CENSZEJFSA-N [(6s,8s,9r,10s,11s,13s,14s,16r,17r)-17-(chloromethylsulfanylcarbonyl)-6,9-difluoro-11-hydroxy-10,13,16-trimethyl-3-oxo-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl] propanoate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCCl)(OC(=O)CC)[C@@]2(C)C[C@@H]1O MVLBCBPGBUAVJQ-CENSZEJFSA-N 0.000 description 1
- JXWVQHSDWAODPY-HHJIKABBSA-N [(6s,8s,9s,10r,11s,13s,14s,17r)-6-fluoro-11-hydroxy-17-(2-hydroxyacetyl)-10,13-dimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-17-yl] pentanoate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@@](C(=O)CO)(OC(=O)CCCC)[C@@]2(C)C[C@@H]1O JXWVQHSDWAODPY-HHJIKABBSA-N 0.000 description 1
- FMEKKTLRKMEQKJ-RCJJHWPHSA-N [(7r,8s,9s,13s,14s,17r)-1-benzoyloxy-17-ethynyl-17-hydroxy-7,13-dimethyl-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-3-yl] benzoate Chemical compound C([C@H]([C@H]1[C@H]2[C@]([C@](CC2)(O)C#C)(C)CC[C@@H]1C1=C(OC(=O)C=2C=CC=CC=2)C=2)C)C1=CC=2OC(=O)C1=CC=CC=C1 FMEKKTLRKMEQKJ-RCJJHWPHSA-N 0.000 description 1
- WWSKHPDYSWDMNC-YRNSVOBJSA-N [(8r,9s,10r,13s,14s,16r,17r)-17-acetyl-6-chloro-10,13,16-trimethyl-3-oxo-2,8,9,11,12,14,15,16-octahydro-1h-cyclopenta[a]phenanthren-17-yl] acetate Chemical compound C1=C(Cl)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(C)=O)(OC(C)=O)[C@@]1(C)CC2 WWSKHPDYSWDMNC-YRNSVOBJSA-N 0.000 description 1
- AHMMSNQYOPMLSX-CNQKSJKFSA-N [(8r,9s,10r,13s,14s,17s)-10,13-dimethyl-3-oxo-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-17-yl] undec-10-enoate Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)OC(=O)CCCCCCCCC=C)[C@@H]4[C@@H]3CCC2=C1 AHMMSNQYOPMLSX-CNQKSJKFSA-N 0.000 description 1
- GACKBTMUYYITHY-OLPLGMLDSA-N [(8r,9s,10r,13s,14s,17s)-6-fluoro-10,13-dimethyl-3-oxo-1,2,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl] propanoate Chemical compound C1C(F)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CC)[C@@]1(C)CC2 GACKBTMUYYITHY-OLPLGMLDSA-N 0.000 description 1
- FXMSQUVSUXBBAB-TXDQRGGKSA-N [(8r,9s,13s,14s,17s)-13-methyl-3-propanoyloxy-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-17-yl] pyridine-3-carboxylate Chemical compound O([C@@H]1[C@@]2(C)CC[C@@H]3C4=CC=C(C=C4CC[C@H]3[C@@H]2CC1)OC(=O)CC)C(=O)C1=CC=CN=C1 FXMSQUVSUXBBAB-TXDQRGGKSA-N 0.000 description 1
- FPVRUILUEYSIMD-RPRRAYFGSA-N [(8s,9r,10s,11s,13s,14s,16r,17r)-9-fluoro-11-hydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-3-oxo-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl] acetate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(OC(C)=O)[C@@]1(C)C[C@@H]2O FPVRUILUEYSIMD-RPRRAYFGSA-N 0.000 description 1
- AEVUURWLDCELOV-AYVPJYCDSA-N [(9s,13s,14s,17s)-17-(furan-3-yl)-17-hydroxy-13-methyl-9,11,12,14,15,16-hexahydro-6h-cyclopenta[a]phenanthren-3-yl] acetate Chemical compound C=1([C@]2(O)CC[C@@H]3[C@]2(C)CC[C@@H]2C4=CC=C(C=C4CC=C23)OC(=O)C)C=COC=1 AEVUURWLDCELOV-AYVPJYCDSA-N 0.000 description 1
- JGKRSFYTHYYJNX-ONNFQVAWSA-N [(e)-(5-cyanothiophen-2-yl)methylideneamino]thiourea Chemical compound NC(=S)N\N=C\C1=CC=C(C#N)S1 JGKRSFYTHYYJNX-ONNFQVAWSA-N 0.000 description 1
- QTPSNPBHOMYPHQ-NKXVUIFGSA-N [(e)-3-phenylprop-2-enyl] (2s,4as,6ar,6as,6br,8ar,10s,12as,14br)-10-acetyloxy-2,4a,6a,6b,9,9,12a-heptamethyl-13-oxo-3,4,5,6,6a,7,8,8a,10,11,12,14b-dodecahydro-1h-picene-2-carboxylate Chemical compound O=C([C@]1(C)C[C@H]2C3=CC(=O)[C@H]4[C@@]([C@@]3(CC[C@@]2(C)CC1)C)(C)CC[C@@H]1[C@]4(C)CC[C@@H](C1(C)C)OC(=O)C)OC\C=C\C1=CC=CC=C1 QTPSNPBHOMYPHQ-NKXVUIFGSA-N 0.000 description 1
- FSAVDKDHPDSCTO-WQLSENKSSA-N [(z)-2-chloro-1-(2,4-dichlorophenyl)ethenyl] diethyl phosphate Chemical compound CCOP(=O)(OCC)O\C(=C/Cl)C1=CC=C(Cl)C=C1Cl FSAVDKDHPDSCTO-WQLSENKSSA-N 0.000 description 1
- ARIFZLJIERKKEL-SNNHKCHZSA-N [(z)-[(1e)-1-(carbamothioylhydrazinylidene)-3-ethoxybutan-2-ylidene]amino]thiourea Chemical compound CCOC(C)\C(=N/NC(N)=S)\C=N\NC(N)=S ARIFZLJIERKKEL-SNNHKCHZSA-N 0.000 description 1
- XPHBRTNHVJSEQD-ATJXCDBQSA-N [(z)-[3-(diethylamino)-1-phenylpropylidene]amino] n-(4-methoxyphenyl)carbamate Chemical compound C=1C=CC=CC=1C(/CCN(CC)CC)=N\OC(=O)NC1=CC=C(OC)C=C1 XPHBRTNHVJSEQD-ATJXCDBQSA-N 0.000 description 1
- YOKPRDAUBGOISU-UHFFFAOYSA-N [1-(3-methylbutoxy)-3-morpholin-4-ylpropan-2-yl] 3,4,5-trimethoxybenzoate Chemical compound COC1=C(OC)C(OC)=CC(C(=O)OC(COCCC(C)C)CN2CCOCC2)=C1 YOKPRDAUBGOISU-UHFFFAOYSA-N 0.000 description 1
- JAHOJXDGNNONSL-UHFFFAOYSA-N [1-(4-methylpiperazin-1-yl)-3-(2-methylpropoxy)propan-2-yl] 2-phenylbutanoate Chemical compound C=1C=CC=CC=1C(CC)C(=O)OC(COCC(C)C)CN1CCN(C)CC1 JAHOJXDGNNONSL-UHFFFAOYSA-N 0.000 description 1
- QJGVXJYGDBSPSJ-UHFFFAOYSA-N [1-(6,7-dimethoxyphthalazin-1-yl)piperidin-4-yl] n-ethylcarbamate Chemical compound C1CC(OC(=O)NCC)CCN1C1=NN=CC2=CC(OC)=C(OC)C=C12 QJGVXJYGDBSPSJ-UHFFFAOYSA-N 0.000 description 1
- KMLCRELJHYKIIL-UHFFFAOYSA-N [1-(azanidylmethyl)cyclohexyl]methylazanide;platinum(2+);sulfuric acid Chemical compound [Pt+2].OS(O)(=O)=O.[NH-]CC1(C[NH-])CCCCC1 KMLCRELJHYKIIL-UHFFFAOYSA-N 0.000 description 1
- RVPQELWEPXXXPC-UHFFFAOYSA-N [1-[4-(4-fluorophenyl)-4-oxobutyl]-3,4,4a,5,6,7,8,8a-octahydro-2h-quinolin-4-yl] carbamate Chemical compound C12CCCCC2C(OC(=O)N)CCN1CCCC(=O)C1=CC=C(F)C=C1 RVPQELWEPXXXPC-UHFFFAOYSA-N 0.000 description 1
- GWZZGHHMOLPYSZ-VMPITWQZSA-N [1-[4-methyl-7-(morpholine-4-carbonylamino)-2-oxochromen-3-yl]-3-morpholin-4-ylpropan-2-yl] (e)-3-(3,4,5-trimethoxyphenyl)prop-2-enoate Chemical compound COC1=C(OC)C(OC)=CC(\C=C\C(=O)OC(CN2CCOCC2)CC=2C(OC3=CC(NC(=O)N4CCOCC4)=CC=C3C=2C)=O)=C1 GWZZGHHMOLPYSZ-VMPITWQZSA-N 0.000 description 1
- FBRAWBYQGRLCEK-UHFFFAOYSA-N [17-(2-chloroacetyl)-9-fluoro-10,13,16-trimethyl-3,11-dioxo-7,8,12,14,15,16-hexahydro-6h-cyclopenta[a]phenanthren-17-yl] butanoate Chemical compound C1CC2=CC(=O)C=CC2(C)C2(F)C1C1CC(C)C(C(=O)CCl)(OC(=O)CCC)C1(C)CC2=O FBRAWBYQGRLCEK-UHFFFAOYSA-N 0.000 description 1
- YHEMKMZUJKPOCO-UHFFFAOYSA-N [2-(2,6-dichloroanilino)-4,5-dihydroimidazol-1-yl]-phenylmethanone Chemical compound ClC1=CC=CC(Cl)=C1NC1=NCCN1C(=O)C1=CC=CC=C1 YHEMKMZUJKPOCO-UHFFFAOYSA-N 0.000 description 1
- UDBQGHIODMCIFZ-UHFFFAOYSA-N [2-(2-methyl-5-pyridin-3-yl-1,2,4-triazol-3-yl)phenyl]methanol Chemical compound CN1N=C(C=2C=NC=CC=2)N=C1C1=CC=CC=C1CO UDBQGHIODMCIFZ-UHFFFAOYSA-N 0.000 description 1
- AVXDZNOJIVOGRR-UHFFFAOYSA-N [2-(diethylamino)-1-phenylethyl] benzoate Chemical compound C=1C=CC=CC=1C(CN(CC)CC)OC(=O)C1=CC=CC=C1 AVXDZNOJIVOGRR-UHFFFAOYSA-N 0.000 description 1
- BEECRKDUIIQEBI-UHFFFAOYSA-N [2-(phenylcarbamoyl)phenyl] n-methylcarbamate Chemical compound CNC(=O)OC1=CC=CC=C1C(=O)NC1=CC=CC=C1 BEECRKDUIIQEBI-UHFFFAOYSA-N 0.000 description 1
- XZSRRNFBEIOBDA-CFNBKWCHSA-N [2-[(2s,4s)-4-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-3,4-dihydro-1h-tetracen-2-yl]-2-oxoethyl] 2,2-diethoxyacetate Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)C(OCC)OCC)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 XZSRRNFBEIOBDA-CFNBKWCHSA-N 0.000 description 1
- ICKMASVVMCGZLR-UHFFFAOYSA-N [2-[(4-chlorophenyl)carbamoyl]-4,6-diiodophenyl] acetate Chemical compound CC(=O)OC1=C(I)C=C(I)C=C1C(=O)NC1=CC=C(Cl)C=C1 ICKMASVVMCGZLR-UHFFFAOYSA-N 0.000 description 1
- RACDDTQBAFEERP-PLTZVPCUSA-N [2-[(6s,8s,9s,10r,13s,14s,17r)-6-chloro-17-hydroxy-10,13-dimethyl-3,11-dioxo-6,7,8,9,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl]-2-oxoethyl] acetate Chemical compound C1([C@@H](Cl)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@@](C(=O)COC(=O)C)(O)[C@@]2(C)CC1=O RACDDTQBAFEERP-PLTZVPCUSA-N 0.000 description 1
- HLAFSNJRKZLMPT-UHFFFAOYSA-N [2-[2-(aminomethyl)phenyl]sulfanylphenyl]methanol Chemical compound NCC1=CC=CC=C1SC1=CC=CC=C1CO HLAFSNJRKZLMPT-UHFFFAOYSA-N 0.000 description 1
- OXCRVQDEVWYAJE-UHFFFAOYSA-N [2-[4-(diethylamino)butylcarbamoyl]phenyl] acetate Chemical compound CCN(CC)CCCCNC(=O)C1=CC=CC=C1OC(C)=O OXCRVQDEVWYAJE-UHFFFAOYSA-N 0.000 description 1
- NRFGEDASJHBPPN-UHFFFAOYSA-N [2-bromo-6-[(4-bromophenyl)carbamothioyl]-4-chlorophenyl] acetate Chemical compound CC(=O)OC1=C(Br)C=C(Cl)C=C1C(=S)NC1=CC=C(Br)C=C1 NRFGEDASJHBPPN-UHFFFAOYSA-N 0.000 description 1
- ZMXGPGZXYJCCHZ-UHFFFAOYSA-N [3-(1,3-dimethyl-2,6-dioxopurin-7-yl)-2-(pyridine-3-carbonyloxy)propyl] pyridine-3-carboxylate Chemical compound C1=2C(=O)N(C)C(=O)N(C)C=2N=CN1CC(OC(=O)C=1C=NC=CC=1)COC(=O)C1=CC=CN=C1 ZMXGPGZXYJCCHZ-UHFFFAOYSA-N 0.000 description 1
- FNDSQXMSCAOERP-UHFFFAOYSA-N [3-(2-phenylbutanoyloxy)-2,2-bis(2-phenylbutanoyloxymethyl)propyl] 2-phenylbutanoate Chemical compound C=1C=CC=CC=1C(CC)C(=O)OCC(COC(=O)C(CC)C=1C=CC=CC=1)(COC(=O)C(CC)C=1C=CC=CC=1)COC(=O)C(CC)C1=CC=CC=C1 FNDSQXMSCAOERP-UHFFFAOYSA-N 0.000 description 1
- VJJOKWOJJZKXNV-UHFFFAOYSA-N [3-[2-(tert-butylamino)-1-hydroxyethyl]-5-(2,2-dimethylpropanoyloxy)phenyl] 2,2-dimethylpropanoate Chemical compound CC(C)(C)NCC(O)C1=CC(OC(=O)C(C)(C)C)=CC(OC(=O)C(C)(C)C)=C1 VJJOKWOJJZKXNV-UHFFFAOYSA-N 0.000 description 1
- YRPPAQRAYJVJOS-UHFFFAOYSA-N [3-acetyloxy-2-[[3,5-bis(trifluoromethyl)phenyl]carbamoyl]-4-nitrophenyl] acetate Chemical compound CC(=O)OC1=CC=C([N+]([O-])=O)C(OC(C)=O)=C1C(=O)NC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 YRPPAQRAYJVJOS-UHFFFAOYSA-N 0.000 description 1
- ZINCPWWBSRSXBH-UHFFFAOYSA-N [4-(4-bromophenyl)-1-[4-(4-fluorophenyl)-4-oxobutyl]piperidin-4-yl] decanoate Chemical compound C1CC(OC(=O)CCCCCCCCC)(C=2C=CC(Br)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 ZINCPWWBSRSXBH-UHFFFAOYSA-N 0.000 description 1
- WWXBHTZSYYGCSG-UHFFFAOYSA-N [4-(carbamoylamino)phenyl]arsonic acid Chemical compound NC(=O)NC1=CC=C([As](O)(O)=O)C=C1 WWXBHTZSYYGCSG-UHFFFAOYSA-N 0.000 description 1
- GRALFSQRIBJAHX-UHFFFAOYSA-N [4-(diethylamino)-3-methylbutan-2-yl] 4-(2-methylpropoxy)benzoate Chemical compound CCN(CC)CC(C)C(C)OC(=O)C1=CC=C(OCC(C)C)C=C1 GRALFSQRIBJAHX-UHFFFAOYSA-N 0.000 description 1
- VUDMUAVGKIZQRY-WCWDXBQESA-N [4-[(e)-4-[4-(furan-2-carbonyloxy)phenyl]hex-3-en-3-yl]phenyl] furan-2-carboxylate Chemical compound C=1C=C(OC(=O)C=2OC=CC=2)C=CC=1C(/CC)=C(\CC)C(C=C1)=CC=C1OC(=O)C1=CC=CO1 VUDMUAVGKIZQRY-WCWDXBQESA-N 0.000 description 1
- SALPXBRLSNRTNE-RUDMXATFSA-N [4-[(e)-but-2-en-2-yl]phenyl] acetate Chemical compound C\C=C(/C)C1=CC=C(OC(C)=O)C=C1 SALPXBRLSNRTNE-RUDMXATFSA-N 0.000 description 1
- VZMLEMYJUIIHNF-DQRAZIAOSA-N [4-[(z)-4-(4-propanoyloxyphenyl)hex-3-en-3-yl]phenyl] propanoate Chemical compound C1=CC(OC(=O)CC)=CC=C1C(\CC)=C(\CC)C1=CC=C(OC(=O)CC)C=C1 VZMLEMYJUIIHNF-DQRAZIAOSA-N 0.000 description 1
- SNNLEHVMRDCTNG-UHFFFAOYSA-N [4-[2-(diethylamino)ethoxy]phenyl]-(2-ethyl-1-benzofuran-3-yl)methanone Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(=O)C1=C(CC)OC2=CC=CC=C12 SNNLEHVMRDCTNG-UHFFFAOYSA-N 0.000 description 1
- NRTGWAAGLRTUJZ-UHFFFAOYSA-N [4-[3-(dibutylamino)propoxy]phenyl]-(2-ethylindolizin-3-yl)methanone Chemical compound C1=CC(OCCCN(CCCC)CCCC)=CC=C1C(=O)C1=C(CC)C=C2N1C=CC=C2 NRTGWAAGLRTUJZ-UHFFFAOYSA-N 0.000 description 1
- LKJGKLIBASNLKR-NILNNKIFSA-N [4-[3-[4-[4-[(8r,9s,10s,13r,14s,17r)-10,13-dimethyl-3,7,12-trioxo-1,2,4,5,6,8,9,11,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl]pentanoyloxy]phenyl]-2-oxo-1h-indol-3-yl]phenyl] 4-[(8r,9s,10s,13r,14s,17r)-10,13-dimethyl-3,7,12-trioxo-1,2,4,5,6,8,9 Chemical compound C([C@]1(C)[C@H]2CC(=O)[C@]34C)CC(=O)CC1CC(=O)[C@H]2[C@@H]4CC[C@@H]3C(C)CCC(=O)OC1=CC=C(C2(C3=CC=CC=C3NC2=O)C=2C=CC(OC(=O)CCC(C)[C@@H]3[C@]4(C(=O)C[C@H]5[C@@H](C(CC6CC(=O)CC[C@@]65C)=O)[C@@H]4CC3)C)=CC=2)C=C1 LKJGKLIBASNLKR-NILNNKIFSA-N 0.000 description 1
- JOOSFXXMIOXKAZ-UHFFFAOYSA-H [Au+3].[Au+3].[O-]C(=O)CC(S)C([O-])=O.[O-]C(=O)CC(S)C([O-])=O.[O-]C(=O)CC(S)C([O-])=O Chemical compound [Au+3].[Au+3].[O-]C(=O)CC(S)C([O-])=O.[O-]C(=O)CC(S)C([O-])=O.[O-]C(=O)CC(S)C([O-])=O JOOSFXXMIOXKAZ-UHFFFAOYSA-H 0.000 description 1
- MPLNGQBULSHWQW-IDDKEARESA-M [Br-].C[N+]1(CC(=O)c2ccc(cc2)c3ccccc3)[C@@H]4CC[C@H]1C[C@H](C4)OC(=O)[C@H](CO)c5ccccc5 Chemical compound [Br-].C[N+]1(CC(=O)c2ccc(cc2)c3ccccc3)[C@@H]4CC[C@H]1C[C@H](C4)OC(=O)[C@H](CO)c5ccccc5 MPLNGQBULSHWQW-IDDKEARESA-M 0.000 description 1
- VZIKPPVTSPAWEC-UHFFFAOYSA-N [[amino-[(n'-benzylcarbamimidoyl)amino]methylidene]amino]phosphonic acid Chemical compound OP(=O)(O)NC(/N)=N\C(=N)NCC1=CC=CC=C1 VZIKPPVTSPAWEC-UHFFFAOYSA-N 0.000 description 1
- JURAJLFHWXNPHG-UHFFFAOYSA-N [acetyl(methylcarbamoyl)amino] n-methylcarbamate Chemical compound CNC(=O)ON(C(C)=O)C(=O)NC JURAJLFHWXNPHG-UHFFFAOYSA-N 0.000 description 1
- VJAKOOPUXKMONS-UHFFFAOYSA-N [amino-(2,6-dichloroanilino)methylidene]urea Chemical compound NC(=O)\N=C(/N)NC1=C(Cl)C=CC=C1Cl VJAKOOPUXKMONS-UHFFFAOYSA-N 0.000 description 1
- CWUDZUQQZDXUHF-UHFFFAOYSA-N [cyano-(3-phenoxyphenyl)methyl] 2,2-dimethylspiro[cyclopropane-3,1'-indene]-1-carboxylate Chemical compound C1=CC2=CC=CC=C2C11C(C)(C)C1C(=O)OC(C#N)C(C=1)=CC=CC=1OC1=CC=CC=C1 CWUDZUQQZDXUHF-UHFFFAOYSA-N 0.000 description 1
- DFFCSGGDLGVQFC-UHFFFAOYSA-O [cyclohex-3-en-1-yl(hydroxy)methyl]-hydroxy-oxophosphanium Chemical compound O[P+](=O)C(O)C1CCC=CC1 DFFCSGGDLGVQFC-UHFFFAOYSA-O 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 1
- 229960004373 acetylcholine Drugs 0.000 description 1
- 102000034337 acetylcholine receptors Human genes 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- FFINMCNLQNTKLU-UHFFFAOYSA-N adipiodone Chemical compound OC(=O)C1=C(I)C=C(I)C(NC(=O)CCCCC(=O)NC=2C(=C(C(O)=O)C(I)=CC=2I)I)=C1I FFINMCNLQNTKLU-UHFFFAOYSA-N 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 229940102884 adrenalin Drugs 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- OENHQHLEOONYIE-UKMVMLAPSA-N all-trans beta-carotene Natural products CC=1CCCC(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C OENHQHLEOONYIE-UKMVMLAPSA-N 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical compound C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- 239000002160 alpha blocker Substances 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229940124308 alpha-adrenoreceptor antagonist Drugs 0.000 description 1
- PCRDIRUKXOTDNN-UHFFFAOYSA-L aluminum;sodium;hydrogen carbonate;hydroxide Chemical compound [OH-].[Na+].[Al+3].OC([O-])=O PCRDIRUKXOTDNN-UHFFFAOYSA-L 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 229950002414 amfecloral Drugs 0.000 description 1
- VBZDETYCYXPOAK-OVCLIPMQSA-N amfecloral Chemical compound ClC(Cl)(Cl)/C=N/C(C)CC1=CC=CC=C1 VBZDETYCYXPOAK-OVCLIPMQSA-N 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- JKOQGQFVAUAYPM-UHFFFAOYSA-N amifostine Chemical compound NCCCNCCSP(O)(O)=O JKOQGQFVAUAYPM-UHFFFAOYSA-N 0.000 description 1
- 229960001097 amifostine Drugs 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- 229960004909 aminosalicylic acid Drugs 0.000 description 1
- FRQGJOFRWIILCX-UHFFFAOYSA-N aminoxytriphene Chemical compound C1=CC(OC)=CC=C1C(CN(C)C)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 FRQGJOFRWIILCX-UHFFFAOYSA-N 0.000 description 1
- 229950009931 aminoxytriphene Drugs 0.000 description 1
- 229960002980 amitriptyline oxide Drugs 0.000 description 1
- ZPMKQFOGINQDAM-UHFFFAOYSA-N amitriptylinoxide Chemical compound C1CC2=CC=CC=C2C(=CCC[N+](C)([O-])C)C2=CC=CC=C21 ZPMKQFOGINQDAM-UHFFFAOYSA-N 0.000 description 1
- 229950001993 amixetrine Drugs 0.000 description 1
- ISRODTBNJUAWEJ-UHFFFAOYSA-N amixetrine Chemical compound C=1C=CC=CC=1C(OCCC(C)C)CN1CCCC1 ISRODTBNJUAWEJ-UHFFFAOYSA-N 0.000 description 1
- 229960003731 amlexanox Drugs 0.000 description 1
- SGRYPYWGNKJSDL-UHFFFAOYSA-N amlexanox Chemical compound NC1=C(C(O)=O)C=C2C(=O)C3=CC(C(C)C)=CC=C3OC2=N1 SGRYPYWGNKJSDL-UHFFFAOYSA-N 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 1
- 229960005143 amobarbital sodium Drugs 0.000 description 1
- 229960001444 amodiaquine Drugs 0.000 description 1
- 229950010701 amogastrin Drugs 0.000 description 1
- 108700042255 amogastrin Proteins 0.000 description 1
- 229950009452 amolanone Drugs 0.000 description 1
- HPITVGRITATAFY-UHFFFAOYSA-N amolanone Chemical compound O=C1OC2=CC=CC=C2C1(CCN(CC)CC)C1=CC=CC=C1 HPITVGRITATAFY-UHFFFAOYSA-N 0.000 description 1
- 229960003173 amoproxan Drugs 0.000 description 1
- 229960003204 amorolfine Drugs 0.000 description 1
- LVEXHFZHOIWIIP-UHFFFAOYSA-N amosulalol Chemical compound COC1=CC=CC=C1OCCNCC(O)C1=CC=C(C)C(S(N)(=O)=O)=C1 LVEXHFZHOIWIIP-UHFFFAOYSA-N 0.000 description 1
- 229950010351 amosulalol Drugs 0.000 description 1
- QWGDMFLQWFTERH-UHFFFAOYSA-N amoxapine Chemical compound C12=CC(Cl)=CC=C2OC2=CC=CC=C2N=C1N1CCNCC1 QWGDMFLQWFTERH-UHFFFAOYSA-N 0.000 description 1
- 229960002519 amoxapine Drugs 0.000 description 1
- 229950004281 amoxecaine Drugs 0.000 description 1
- 229950003602 amoxydramine camsilate Drugs 0.000 description 1
- NNAIYOXJNVGUOM-UHFFFAOYSA-N amperozide Chemical compound C1CN(C(=O)NCC)CCN1CCCC(C=1C=CC(F)=CC=1)C1=CC=C(F)C=C1 NNAIYOXJNVGUOM-UHFFFAOYSA-N 0.000 description 1
- 229950000388 amperozide Drugs 0.000 description 1
- ZVSGUZQJNXHNIL-UHFFFAOYSA-N amphenidone Chemical compound NC1=CC=CC(N2C(C=CC=C2)=O)=C1 ZVSGUZQJNXHNIL-UHFFFAOYSA-N 0.000 description 1
- 229950008169 amphenidone Drugs 0.000 description 1
- 229940025084 amphetamine Drugs 0.000 description 1
- GIIGAFADHXYOPM-UHFFFAOYSA-N amphotalide Chemical compound C1=CC(N)=CC=C1OCCCCCN1C(=O)C2=CC=CC=C2C1=O GIIGAFADHXYOPM-UHFFFAOYSA-N 0.000 description 1
- 229950008495 amphotalide Drugs 0.000 description 1
- LSNWBKACGXCGAJ-UHFFFAOYSA-N ampiroxicam Chemical compound CN1S(=O)(=O)C2=CC=CC=C2C(OC(C)OC(=O)OCC)=C1C(=O)NC1=CC=CC=N1 LSNWBKACGXCGAJ-UHFFFAOYSA-N 0.000 description 1
- 229950011249 ampiroxicam Drugs 0.000 description 1
- 229960003683 amprolium Drugs 0.000 description 1
- 229950001528 ampyrimine Drugs 0.000 description 1
- UUINNXPPLPDRQX-UHFFFAOYSA-N ampyzine Chemical compound CN(C)C1=CN=CC=N1 UUINNXPPLPDRQX-UHFFFAOYSA-N 0.000 description 1
- 229950004525 ampyzine Drugs 0.000 description 1
- 229950011291 amquinate Drugs 0.000 description 1
- 229960002105 amrinone Drugs 0.000 description 1
- RNLQIBCLLYYYFJ-UHFFFAOYSA-N amrinone Chemical compound N1C(=O)C(N)=CC(C=2C=CN=CC=2)=C1 RNLQIBCLLYYYFJ-UHFFFAOYSA-N 0.000 description 1
- 229940089837 amygdalin Drugs 0.000 description 1
- YZLOSXFCSIDECK-UHFFFAOYSA-N amygdalin Natural products OCC1OC(OCC2OC(O)C(O)C(O)C2O)C(O)C(O)C1OC(C#N)c3ccccc3 YZLOSXFCSIDECK-UHFFFAOYSA-N 0.000 description 1
- 229960003116 amyl nitrite Drugs 0.000 description 1
- 229960005213 amylmetacresol Drugs 0.000 description 1
- CKGWFZQGEQJZIL-UHFFFAOYSA-N amylmetacresol Chemical compound CCCCCC1=CC=C(C)C=C1O CKGWFZQGEQJZIL-UHFFFAOYSA-N 0.000 description 1
- 229940124326 anaesthetic agent Drugs 0.000 description 1
- KDLNOQQQEBKBQM-DICPTYMLSA-N anagestone acetate Chemical compound C([C@@]12C)CCC=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 KDLNOQQQEBKBQM-DICPTYMLSA-N 0.000 description 1
- 229950002552 anagestone acetate Drugs 0.000 description 1
- 229960001694 anagrelide Drugs 0.000 description 1
- OTBXOEAOVRKTNQ-UHFFFAOYSA-N anagrelide Chemical compound N1=C2NC(=O)CN2CC2=C(Cl)C(Cl)=CC=C21 OTBXOEAOVRKTNQ-UHFFFAOYSA-N 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 229950001003 anaxirone Drugs 0.000 description 1
- YFFNPIAPUDUYAU-UHFFFAOYSA-N anazocine Chemical compound C1N(C)CC2CCCC1C2(OC)C1=CC=CC=C1 YFFNPIAPUDUYAU-UHFFFAOYSA-N 0.000 description 1
- 229950002862 anazocine Drugs 0.000 description 1
- QFVHZQCOUORWEI-ZQHSETAFSA-N anazolene Chemical compound OC1=C2C(=CC(=C1)S(O)(=O)=O)C=C(C=C2\N=N\C1=CC=C(NC2=CC=CC=C2)C2=C(C=CC=C12)S(O)(=O)=O)S(O)(=O)=O QFVHZQCOUORWEI-ZQHSETAFSA-N 0.000 description 1
- 229950004368 anazolene Drugs 0.000 description 1
- XBRNQRFNEAHCPR-UHFFFAOYSA-N ancarolol Chemical compound CC(C)(C)NCC(O)COC1=CC=CC=C1NC(=O)C1=CC=CO1 XBRNQRFNEAHCPR-UHFFFAOYSA-N 0.000 description 1
- 229950006766 ancarolol Drugs 0.000 description 1
- BBDAGFIXKZCXAH-CCXZUQQUSA-N ancitabine Chemical compound N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 BBDAGFIXKZCXAH-CCXZUQQUSA-N 0.000 description 1
- 229950000242 ancitabine Drugs 0.000 description 1
- 229940011037 anethole Drugs 0.000 description 1
- 108010072788 angiogenin Proteins 0.000 description 1
- 229950006323 angiotensin ii Drugs 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 229950004733 anidoxime Drugs 0.000 description 1
- 229950007166 anilamate Drugs 0.000 description 1
- LKYQLAWMNBFNJT-UHFFFAOYSA-N anileridine Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCC1=CC=C(N)C=C1 LKYQLAWMNBFNJT-UHFFFAOYSA-N 0.000 description 1
- 229960002512 anileridine Drugs 0.000 description 1
- 229940039407 aniline Drugs 0.000 description 1
- GNCHTURXQMPGMG-UHFFFAOYSA-N anilopam Chemical compound CC1CC2=CC(OC)=CC=C2CCN1CCC1=CC=C(N)C=C1 GNCHTURXQMPGMG-UHFFFAOYSA-N 0.000 description 1
- 229950006347 anilopam Drugs 0.000 description 1
- PHFDAOXXIZOUIX-UHFFFAOYSA-N anipamil Chemical compound C=1C=CC(OC)=CC=1C(CCCCCCCCCCCC)(C#N)CCCN(C)CCC1=CC=CC(OC)=C1 PHFDAOXXIZOUIX-UHFFFAOYSA-N 0.000 description 1
- 229950011530 anipamil Drugs 0.000 description 1
- 229960000793 aniracetam Drugs 0.000 description 1
- ZXNRTKGTQJPIJK-UHFFFAOYSA-N aniracetam Chemical compound C1=CC(OC)=CC=C1C(=O)N1C(=O)CCC1 ZXNRTKGTQJPIJK-UHFFFAOYSA-N 0.000 description 1
- 229950004699 anirolac Drugs 0.000 description 1
- 229960000613 anisacril Drugs 0.000 description 1
- XRCFXMGQEVUZFC-UHFFFAOYSA-N anisindione Chemical compound C1=CC(OC)=CC=C1C1C(=O)C2=CC=CC=C2C1=O XRCFXMGQEVUZFC-UHFFFAOYSA-N 0.000 description 1
- 229960002138 anisindione Drugs 0.000 description 1
- 229950003530 anisopirol Drugs 0.000 description 1
- HDNJXZZJFPCFHG-UHFFFAOYSA-N anitrazafen Chemical compound C1=CC(OC)=CC=C1C1=NN=C(C)N=C1C1=CC=C(OC)C=C1 HDNJXZZJFPCFHG-UHFFFAOYSA-N 0.000 description 1
- 229950002412 anitrazafen Drugs 0.000 description 1
- 229950000969 anpirtoline Drugs 0.000 description 1
- JDQWJVUVMKPZLU-UHFFFAOYSA-N ansoxetine Chemical compound C=1C=CC=CC=1C(CCN(C)C)OC(C=C1C(=O)C=2)=CC=C1OC=2C1=CC=CC=C1 JDQWJVUVMKPZLU-UHFFFAOYSA-N 0.000 description 1
- 229950004471 ansoxetine Drugs 0.000 description 1
- 229950006351 antafenite Drugs 0.000 description 1
- 229960002469 antazoline Drugs 0.000 description 1
- REYFJDPCWQRWAA-UHFFFAOYSA-N antazoline Chemical compound N=1CCNC=1CN(C=1C=CC=CC=1)CC1=CC=CC=C1 REYFJDPCWQRWAA-UHFFFAOYSA-N 0.000 description 1
- 229950009062 antazonite Drugs 0.000 description 1
- VQQSDVBOXQHCHU-SKPOXZENSA-N antelmycin Chemical compound O[C@@H]1[C@@H](N)[C@H](O)[C@@H](CO)O[C@H]1O[C@H]([C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO)[C@@H]1[C@@H](N)[C@H](O)[C@@H](O)[C@H](N2C(N=C(N)C=C2)=O)O1 VQQSDVBOXQHCHU-SKPOXZENSA-N 0.000 description 1
- 229950000658 anthiolimine Drugs 0.000 description 1
- NUZWLKWWNNJHPT-UHFFFAOYSA-N anthralin Chemical compound C1C2=CC=CC(O)=C2C(=O)C2=C1C=CC=C2O NUZWLKWWNNJHPT-UHFFFAOYSA-N 0.000 description 1
- VGQOVCHZGQWAOI-HYUHUPJXSA-N anthramycin Chemical compound N1[C@@H](O)[C@@H]2CC(\C=C\C(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-HYUHUPJXSA-N 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000001188 anti-phage Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 230000002785 anti-thrombosis Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229950008895 antienite Drugs 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- 229940075103 antimony Drugs 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 229940026189 antimony potassium tartrate Drugs 0.000 description 1
- VEQOALNAAJBPNY-UHFFFAOYSA-N antipyrine Chemical compound CN1C(C)=CC(=O)N1C1=CC=CC=C1 VEQOALNAAJBPNY-UHFFFAOYSA-N 0.000 description 1
- 239000004019 antithrombin Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 229940121357 antivirals Drugs 0.000 description 1
- 229950004064 antrafenine Drugs 0.000 description 1
- XMQVYNAURODYCQ-SLFBBCNNSA-N apalcillin Chemical compound C1([C@@H](NC(=O)C=2C(=C3N=CC=CC3=NC=2)O)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 XMQVYNAURODYCQ-SLFBBCNNSA-N 0.000 description 1
- 229950001979 apalcillin Drugs 0.000 description 1
- HRWVXKVRSNICJQ-GMJIGYHYSA-N apicycline Chemical compound O=C([C@@]1(O)C(O)=C2[C@@H]([C@](C3=CC=CC(O)=C3C2=O)(C)O)C[C@H]1[C@@H](C=1O)N(C)C)C=1C(=O)NC(C(O)=O)N1CCN(CCO)CC1 HRWVXKVRSNICJQ-GMJIGYHYSA-N 0.000 description 1
- 229950008405 apicycline Drugs 0.000 description 1
- 108010091513 apolipoprotein B receptor Proteins 0.000 description 1
- 229960004046 apomorphine Drugs 0.000 description 1
- VMWNQDUVQKEIOC-CYBMUJFWSA-N apomorphine Chemical compound C([C@H]1N(C)CC2)C3=CC=C(O)C(O)=C3C3=C1C2=CC=C3 VMWNQDUVQKEIOC-CYBMUJFWSA-N 0.000 description 1
- 229950006936 apovincamine Drugs 0.000 description 1
- OZDNDGXASTWERN-UHFFFAOYSA-N apovincamine Natural products C1=CC=C2C(CCN3CCC4)=C5C3C4(CC)C=C(C(=O)OC)N5C2=C1 OZDNDGXASTWERN-UHFFFAOYSA-N 0.000 description 1
- 229960002610 apraclonidine Drugs 0.000 description 1
- IEJXVRYNEISIKR-UHFFFAOYSA-N apraclonidine Chemical compound ClC1=CC(N)=CC(Cl)=C1NC1=NCCN1 IEJXVRYNEISIKR-UHFFFAOYSA-N 0.000 description 1
- XZNUGFQTQHRASN-XQENGBIVSA-N apramycin Chemical compound O([C@H]1O[C@@H]2[C@H](O)[C@@H]([C@H](O[C@H]2C[C@H]1N)O[C@@H]1[C@@H]([C@@H](O)[C@H](N)[C@@H](CO)O1)O)NC)[C@@H]1[C@@H](N)C[C@@H](N)[C@H](O)[C@H]1O XZNUGFQTQHRASN-XQENGBIVSA-N 0.000 description 1
- 229950006334 apramycin Drugs 0.000 description 1
- NZLBHDRPUJLHCE-UHFFFAOYSA-N aprindine Chemical compound C1C2=CC=CC=C2CC1N(CCCN(CC)CC)C1=CC=CC=C1 NZLBHDRPUJLHCE-UHFFFAOYSA-N 0.000 description 1
- 229960004957 aprindine Drugs 0.000 description 1
- 229960003153 aprobarbital Drugs 0.000 description 1
- UORJNBVJVRLXMQ-UHFFFAOYSA-N aprobarbital Chemical compound C=CCC1(C(C)C)C(=O)NC(=O)NC1=O UORJNBVJVRLXMQ-UHFFFAOYSA-N 0.000 description 1
- 229950005147 aprofene Drugs 0.000 description 1
- 229950011611 aptazapine Drugs 0.000 description 1
- MNHDDERDSNZCCK-UHFFFAOYSA-N aptazapine Chemical compound C1C2=CC=CC=C2N2CCN(C)CC2C2=CC=CN21 MNHDDERDSNZCCK-UHFFFAOYSA-N 0.000 description 1
- 229950010659 aptocaine Drugs 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 1
- HXWOWBFXYUFFKS-PSJNWGMYSA-N aranotin Chemical compound C1C2=COC=C[C@H](O)[C@H]2N(C2=O)[C@]31SS[C@]21CC2=COC=C[C@H](OC(=O)C)[C@H]2N1C3=O HXWOWBFXYUFFKS-PSJNWGMYSA-N 0.000 description 1
- 229950001980 aranotin Drugs 0.000 description 1
- HXWOWBFXYUFFKS-UHFFFAOYSA-N aranotin Natural products C1C2=COC=CC(O)C2N(C2=O)C31SSC21CC2=COC=CC(OC(=O)C)C2N1C3=O HXWOWBFXYUFFKS-UHFFFAOYSA-N 0.000 description 1
- 229950002312 arbaprostil Drugs 0.000 description 1
- 229960005397 arbekacin Drugs 0.000 description 1
- MKKYBZZTJQGVCD-XTCKQBCOSA-N arbekacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)CC[C@H]1N MKKYBZZTJQGVCD-XTCKQBCOSA-N 0.000 description 1
- 229950005800 arclofenin Drugs 0.000 description 1
- MSDLMFOYXOFYBK-UHFFFAOYSA-N arclofenin Chemical compound OC(=O)CN(CC(O)=O)CC(=O)NC1=CC=C(Cl)C=C1C(=O)C1=CC=CC=C1 MSDLMFOYXOFYBK-UHFFFAOYSA-N 0.000 description 1
- 229950003131 arfendazam Drugs 0.000 description 1
- NXJWVCHVPUCWJS-UHFFFAOYSA-N arfendazam Chemical compound C12=CC(Cl)=CC=C2N(C(=O)OCC)CCC(=O)N1C1=CC=CC=C1 NXJWVCHVPUCWJS-UHFFFAOYSA-N 0.000 description 1
- KXNPVXPOPUZYGB-XYVMCAHJSA-N argatroban Chemical compound OC(=O)[C@H]1C[C@H](C)CCN1C(=O)[C@H](CCCN=C(N)N)NS(=O)(=O)C1=CC=CC2=C1NC[C@H](C)C2 KXNPVXPOPUZYGB-XYVMCAHJSA-N 0.000 description 1
- 229960003856 argatroban Drugs 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- DIXRMZGIJNJUGL-UHFFFAOYSA-N arildone Chemical compound CCC(=O)C(C(=O)CC)CCCCCCOC1=CC=C(OC)C=C1Cl DIXRMZGIJNJUGL-UHFFFAOYSA-N 0.000 description 1
- 229950003470 arildone Drugs 0.000 description 1
- LAWLHMWODZUZJH-UHFFFAOYSA-N arnolol Chemical compound COCCC1=CC=C(OCC(O)C(C)(C)N)C=C1 LAWLHMWODZUZJH-UHFFFAOYSA-N 0.000 description 1
- 229950005503 arnolol Drugs 0.000 description 1
- 229950002927 aronixil Drugs 0.000 description 1
- BHIAIPWSVYSKJS-UHFFFAOYSA-N arotinolol Chemical compound S1C(SCC(O)CNC(C)(C)C)=NC(C=2SC(=CC=2)C(N)=O)=C1 BHIAIPWSVYSKJS-UHFFFAOYSA-N 0.000 description 1
- 229950010731 arotinolol Drugs 0.000 description 1
- 229950009330 arpromidine Drugs 0.000 description 1
- XKNKHVGWJDPIRJ-UHFFFAOYSA-N arsanilic acid Chemical compound NC1=CC=C([As](O)(O)=O)C=C1 XKNKHVGWJDPIRJ-UHFFFAOYSA-N 0.000 description 1
- 229950002705 arsanilic acid Drugs 0.000 description 1
- MRUDSZSRLQAPOG-UHFFFAOYSA-N arsthinol Chemical compound C1=C(O)C(NC(=O)C)=CC([As]2SC(CO)CS2)=C1 MRUDSZSRLQAPOG-UHFFFAOYSA-N 0.000 description 1
- 229960000778 arsthinol Drugs 0.000 description 1
- 229960004191 artemisinin Drugs 0.000 description 1
- BLUAFEHZUWYNDE-NNWCWBAJSA-N artemisinin Chemical compound C([C@](OO1)(C)O2)C[C@H]3[C@H](C)CC[C@@H]4[C@@]31[C@@H]2OC(=O)[C@@H]4C BLUAFEHZUWYNDE-NNWCWBAJSA-N 0.000 description 1
- 229930101531 artemisinin Natural products 0.000 description 1
- 229960003831 articaine Drugs 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- IORHSKBXWWSQME-UHFFFAOYSA-N asocainol Chemical compound C1C2=CC=C(OC)C(O)=C2C2=CC(OC)=CC=C2CCN(C)C1CCC1=CC=CC=C1 IORHSKBXWWSQME-UHFFFAOYSA-N 0.000 description 1
- 229950003482 asocainol Drugs 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 229960000202 aspoxicillin Drugs 0.000 description 1
- 230000000712 assembly Effects 0.000 description 1
- 238000000429 assembly Methods 0.000 description 1
- GXDALQBWZGODGZ-UHFFFAOYSA-N astemizole Chemical compound C1=CC(OC)=CC=C1CCN1CCC(NC=2N(C3=CC=CC=C3N=2)CC=2C=CC(F)=CC=2)CC1 GXDALQBWZGODGZ-UHFFFAOYSA-N 0.000 description 1
- PEPMWUSGRKINHX-TXTPUJOMSA-N atamestane Chemical compound C1C[C@@H]2[C@@]3(C)C(C)=CC(=O)C=C3CC[C@H]2[C@@H]2CCC(=O)[C@]21C PEPMWUSGRKINHX-TXTPUJOMSA-N 0.000 description 1
- 229950004810 atamestane Drugs 0.000 description 1
- HSWPZIDYAHLZDD-UHFFFAOYSA-N atipamezole Chemical compound C1C2=CC=CC=C2CC1(CC)C1=CN=CN1 HSWPZIDYAHLZDD-UHFFFAOYSA-N 0.000 description 1
- 229960003002 atipamezole Drugs 0.000 description 1
- 229950004318 atiprosin Drugs 0.000 description 1
- WXNVFYIBELASJW-QUCCMNQESA-N atiprosin Chemical compound C1=CC=C2N3CC[C@H]4N(C(C)C)CCN(CC)[C@@H]4C3=C(C)C2=C1 WXNVFYIBELASJW-QUCCMNQESA-N 0.000 description 1
- 229950006231 atolide Drugs 0.000 description 1
- 229960001862 atracurium Drugs 0.000 description 1
- 229950008951 atracurium besilate Drugs 0.000 description 1
- XXZSQOVSEBAPGS-UHFFFAOYSA-L atracurium besylate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1.[O-]S(=O)(=O)C1=CC=CC=C1.C1=C(OC)C(OC)=CC=C1CC1[N+](CCC(=O)OCCCCCOC(=O)CC[N+]2(C)C(C3=CC(OC)=C(OC)C=C3CC2)CC=2C=C(OC)C(OC)=CC=2)(C)CCC2=CC(OC)=C(OC)C=C21 XXZSQOVSEBAPGS-UHFFFAOYSA-L 0.000 description 1
- 230000001746 atrial effect Effects 0.000 description 1
- 229950008800 atromepine Drugs 0.000 description 1
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 1
- 229960000396 atropine Drugs 0.000 description 1
- 229950010917 atropine oxyde Drugs 0.000 description 1
- HGWPFSBHDACWNL-LZYIFBDPSA-N atropine oxyde Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)[N+]2([O-])C)C(=O)C(CO)C1=CC=CC=C1 HGWPFSBHDACWNL-LZYIFBDPSA-N 0.000 description 1
- AUJRCFUBUPVWSZ-XTZHGVARSA-M auranofin Chemical compound CCP(CC)(CC)=[Au]S[C@@H]1O[C@H](COC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O AUJRCFUBUPVWSZ-XTZHGVARSA-M 0.000 description 1
- 229960005207 auranofin Drugs 0.000 description 1
- ODENGJOFMCVWCT-UHFFFAOYSA-M aurothioglycanide Chemical compound [Au+].[S-]CC(=O)NC1=CC=CC=C1 ODENGJOFMCVWCT-UHFFFAOYSA-M 0.000 description 1
- 229950002878 aurothioglycanide Drugs 0.000 description 1
- XIRGHRXBGGPPKY-FCNCREMHSA-N avilamycin A Chemical compound O([C@H]1[C@H](O)C[C@@H](O[C@@H]1C)O[C@H]1[C@H](O)CC2(O[C@]3(C)C[C@@H](O[C@H](C)[C@H]3O2)O[C@H]2[C@@H](OC)[C@@H](C)O[C@H]([C@@H]2O)O[C@H]2[C@H](O)[C@H](OC)[C@H](O[C@H]3[C@@H]([C@@H]4O[C@]5(O[C@H]4CO3)[C@@H]3OCO[C@H]3[C@@](O)([C@@H](C)O5)C(C)=O)OC(=O)C(C)C)O[C@@H]2COC)O[C@@H]1C)C(=O)C1=C(C)C(Cl)=C(O)C(Cl)=C1OC XIRGHRXBGGPPKY-FCNCREMHSA-N 0.000 description 1
- WXNRAKRZUCLRBP-UHFFFAOYSA-N avridine Chemical compound CCCCCCCCCCCCCCCCCCN(CCCN(CCO)CCO)CCCCCCCCCCCCCCCCCC WXNRAKRZUCLRBP-UHFFFAOYSA-N 0.000 description 1
- 229950010555 avridine Drugs 0.000 description 1
- 229950001035 axamozide Drugs 0.000 description 1
- 229950003588 axetil Drugs 0.000 description 1
- 229950003139 azabon Drugs 0.000 description 1
- 229950006573 azabuperone Drugs 0.000 description 1
- 229950009743 azaclorzine Drugs 0.000 description 1
- AKNQMEBLVAMSNZ-UHFFFAOYSA-N azaconazole Chemical compound ClC1=CC(Cl)=CC=C1C1(CN2N=CN=C2)OCCO1 AKNQMEBLVAMSNZ-UHFFFAOYSA-N 0.000 description 1
- 229950000294 azaconazole Drugs 0.000 description 1
- FMTFZYKYVZBISL-HUVRVWIJSA-N azacosterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](N(C)CCCN(C)C)[C@@]1(C)CC2 FMTFZYKYVZBISL-HUVRVWIJSA-N 0.000 description 1
- 229950005866 azacosterol Drugs 0.000 description 1
- 229950008193 azacyclonol Drugs 0.000 description 1
- 229950001298 azaftozine Drugs 0.000 description 1
- 229950003696 azaloxan Drugs 0.000 description 1
- CDFTVVPIMLXJRX-HNNXBMFYSA-N azaloxan Chemical compound O=C1NCCN1C1CCN(CC[C@@H]2OC3=CC=CC=C3OC2)CC1 CDFTVVPIMLXJRX-HNNXBMFYSA-N 0.000 description 1
- 229950009336 azamethonium bromide Drugs 0.000 description 1
- FMHQJXGMLMSMLC-WBUYAQKGSA-N azamulin Chemical compound O([C@@H]1C[C@@]([C@H]([C@H](C)[C@]23CCC(=O)[C@H]2[C@@]1(C)[C@@H](CC3)C)O)(C)CC)C(=O)CSC1=NNC(N)=N1 FMHQJXGMLMSMLC-WBUYAQKGSA-N 0.000 description 1
- 229950008762 azamulin Drugs 0.000 description 1
- 229950010669 azanator Drugs 0.000 description 1
- 229960004904 azanidazole Drugs 0.000 description 1
- LHIALLMPKJMSIQ-NSCUHMNNSA-N azanidazole Chemical compound C1=C([N+]([O-])=O)N(C)C(\C=C\C=2N=C(N)N=CC=2)=N1 LHIALLMPKJMSIQ-NSCUHMNNSA-N 0.000 description 1
- BOFZOTMTKBQRAB-UHFFFAOYSA-N azanium;2-carboxyphenolate Chemical compound N.OC(=O)C1=CC=CC=C1O BOFZOTMTKBQRAB-UHFFFAOYSA-N 0.000 description 1
- 229950003616 azaperone Drugs 0.000 description 1
- XTKDAFGWCDAMPY-UHFFFAOYSA-N azaperone Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCN(C=2N=CC=CC=2)CC1 XTKDAFGWCDAMPY-UHFFFAOYSA-N 0.000 description 1
- RKNSPEOBXHFNTD-DQCUJPBYSA-N azaprocin Chemical compound C([C@H]1CC[C@@H](C2)N1C(=O)CC)N2C\C=C\C1=CC=CC=C1 RKNSPEOBXHFNTD-DQCUJPBYSA-N 0.000 description 1
- 229950003771 azaprocin Drugs 0.000 description 1
- 229960001671 azapropazone Drugs 0.000 description 1
- WOIIIUDZSOLAIW-NSHDSACASA-N azapropazone Chemical compound C1=C(C)C=C2N3C(=O)[C@H](CC=C)C(=O)N3C(N(C)C)=NC2=C1 WOIIIUDZSOLAIW-NSHDSACASA-N 0.000 description 1
- 229950001620 azaquinzole Drugs 0.000 description 1
- QQOBRRFOVWGIMD-OJAKKHQRSA-N azaribine Chemical compound CC(=O)O[C@@H]1[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1N1C(=O)NC(=O)C=N1 QQOBRRFOVWGIMD-OJAKKHQRSA-N 0.000 description 1
- 229950010054 azaribine Drugs 0.000 description 1
- 229950011321 azaserine Drugs 0.000 description 1
- PYNSMGBGABAWPB-UHFFFAOYSA-M azaspirium chloride Chemical compound [Cl-].COC1=C2OC=CC2=C(OC)C(C(C=2C3)=O)=C1OC=2C(=C)C[N+]13CCCCC1 PYNSMGBGABAWPB-UHFFFAOYSA-M 0.000 description 1
- 229950011207 azaspirium chloride Drugs 0.000 description 1
- AXLOCHLTNQDFFS-BESJYZOMSA-N azastene Chemical compound C([C@H]1[C@@H]2CC[C@@]([C@]2(CC[C@@H]1[C@@]1(C)C2)C)(O)C)C=C1C(C)(C)C1=C2C=NO1 AXLOCHLTNQDFFS-BESJYZOMSA-N 0.000 description 1
- 229950007499 azastene Drugs 0.000 description 1
- HRXVDDOKERXBEY-UHFFFAOYSA-N azatepa Chemical compound C1CN1P(=O)(N1CC1)N(CC)C1=NN=CS1 HRXVDDOKERXBEY-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960004574 azelastine Drugs 0.000 description 1
- 229950006225 azepexole Drugs 0.000 description 1
- FEJCIXJKPISCJV-UHFFFAOYSA-N azepindole Chemical compound C1CCNCC2=CC3=CC=CC=C3N21 FEJCIXJKPISCJV-UHFFFAOYSA-N 0.000 description 1
- 229950008946 azepindole Drugs 0.000 description 1
- 229960002278 azidamfenicol Drugs 0.000 description 1
- SGRUZFCHLOFYHZ-MWLCHTKSSA-N azidamfenicol Chemical compound [N-]=[N+]=NCC(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 SGRUZFCHLOFYHZ-MWLCHTKSSA-N 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- ODFHGIPNGIAMDK-NJBDSQKTSA-N azidocillin Chemical compound C1([C@@H](N=[N+]=[N-])C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 ODFHGIPNGIAMDK-NJBDSQKTSA-N 0.000 description 1
- 229960004328 azidocillin Drugs 0.000 description 1
- 229950009664 azimexon Drugs 0.000 description 1
- SSLKKMZJCJBOML-UHFFFAOYSA-N azintamide Chemical compound CCN(CC)C(=O)CSC1=CC=C(Cl)N=N1 SSLKKMZJCJBOML-UHFFFAOYSA-N 0.000 description 1
- 229960004174 azintamide Drugs 0.000 description 1
- GMJAPDJBIOQXSW-UHFFFAOYSA-N azipramine Chemical compound C=1C(C2=3)=CC=CC=3CCC3=CC=CC=C3N2C=1CCN(C)CC1=CC=CC=C1 GMJAPDJBIOQXSW-UHFFFAOYSA-N 0.000 description 1
- 229950001502 azipramine Drugs 0.000 description 1
- 150000001541 aziridines Chemical class 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 229960003623 azlocillin Drugs 0.000 description 1
- JTWOMNBEOCYFNV-NFFDBFGFSA-N azlocillin Chemical compound N([C@@H](C(=O)N[C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C=1C=CC=CC=1)C(=O)N1CCNC1=O JTWOMNBEOCYFNV-NFFDBFGFSA-N 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- 229950005181 azolimine Drugs 0.000 description 1
- KPMDCTAAOGEIKO-UHFFFAOYSA-N azolimine Chemical compound N=C1N(C)C(=O)CN1C1=CC=CC=C1 KPMDCTAAOGEIKO-UHFFFAOYSA-N 0.000 description 1
- 229960004988 azosemide Drugs 0.000 description 1
- IIOPLILENRZKRV-UHFFFAOYSA-N azosemide Chemical compound C=1C=CSC=1CNC=1C=C(Cl)C(S(=O)(=O)N)=CC=1C1=NN=N[N]1 IIOPLILENRZKRV-UHFFFAOYSA-N 0.000 description 1
- 229950004295 azotomycin Drugs 0.000 description 1
- 229960003644 aztreonam Drugs 0.000 description 1
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 1
- 229950003578 azumolene Drugs 0.000 description 1
- 229960000794 baclofen Drugs 0.000 description 1
- 229950008796 bacmecillinam Drugs 0.000 description 1
- 229960004168 balsalazide Drugs 0.000 description 1
- IPOKCKJONYRRHP-FMQUCBEESA-N balsalazide Chemical compound C1=CC(C(=O)NCCC(=O)O)=CC=C1\N=N\C1=CC=C(O)C(C(O)=O)=C1 IPOKCKJONYRRHP-FMQUCBEESA-N 0.000 description 1
- XRGNABQSJLQUGV-UHFFFAOYSA-N bamaluzole Chemical compound N1=CC=C2N(C)C=NC2=C1OCC1=CC=CC=C1Cl XRGNABQSJLQUGV-UHFFFAOYSA-N 0.000 description 1
- 229950010457 bamaluzole Drugs 0.000 description 1
- ANZXOIAKUNOVQU-UHFFFAOYSA-N bambuterol Chemical compound CN(C)C(=O)OC1=CC(OC(=O)N(C)C)=CC(C(O)CNC(C)(C)C)=C1 ANZXOIAKUNOVQU-UHFFFAOYSA-N 0.000 description 1
- 229960003060 bambuterol Drugs 0.000 description 1
- 229960004162 bamethan Drugs 0.000 description 1
- RDUHXGIIUDVSHR-UHFFFAOYSA-N bamethan Chemical compound CCCCNCC(O)C1=CC=C(O)C=C1 RDUHXGIIUDVSHR-UHFFFAOYSA-N 0.000 description 1
- VVUYEFBRTFASAH-UHFFFAOYSA-N bamifylline Chemical compound N=1C=2N(C)C(=O)N(C)C(=O)C=2N(CCN(CCO)CC)C=1CC1=CC=CC=C1 VVUYEFBRTFASAH-UHFFFAOYSA-N 0.000 description 1
- 229960005176 bamifylline Drugs 0.000 description 1
- 229960002526 bamipine Drugs 0.000 description 1
- VZSXTYKGYWISGQ-UHFFFAOYSA-N bamipine Chemical compound C1CN(C)CCC1N(C=1C=CC=CC=1)CC1=CC=CC=C1 VZSXTYKGYWISGQ-UHFFFAOYSA-N 0.000 description 1
- 229950005881 bamnidazole Drugs 0.000 description 1
- AIOWJIMWVFWROP-UHFFFAOYSA-N baquiloprim Chemical compound C12=CC=CN=C2C(N(C)C)=C(C)C=C1CC1=CN=C(N)N=C1N AIOWJIMWVFWROP-UHFFFAOYSA-N 0.000 description 1
- 229950004557 baquiloprim Drugs 0.000 description 1
- MJCBWPMBFCUHBP-NPULLEENSA-N barbexaclone Chemical compound CN[C@@H](C)CC1CCCCC1.C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O MJCBWPMBFCUHBP-NPULLEENSA-N 0.000 description 1
- 229960002910 barbexaclone Drugs 0.000 description 1
- 229960002319 barbital Drugs 0.000 description 1
- 229950002921 barucainide Drugs 0.000 description 1
- 229960003513 batilol Drugs 0.000 description 1
- XFILPEOLDIKJHX-QYZOEREBSA-N batimastat Chemical compound C([C@@H](C(=O)NC)NC(=O)[C@H](CC(C)C)[C@H](CSC=1SC=CC=1)C(=O)NO)C1=CC=CC=C1 XFILPEOLDIKJHX-QYZOEREBSA-N 0.000 description 1
- 229950001858 batimastat Drugs 0.000 description 1
- KRNDIPHOJLIHRI-UHFFFAOYSA-N bazinaprine Chemical compound N#CC1=CC(C=2C=CC=CC=2)=NN=C1NCCN1CCOCC1 KRNDIPHOJLIHRI-UHFFFAOYSA-N 0.000 description 1
- 229950005683 bazinaprine Drugs 0.000 description 1
- JPYQFYIEOUVJDU-UHFFFAOYSA-N beclamide Chemical compound ClCCC(=O)NCC1=CC=CC=C1 JPYQFYIEOUVJDU-UHFFFAOYSA-N 0.000 description 1
- 229960005200 beclamide Drugs 0.000 description 1
- YWQGBCXVCXMSLJ-UHFFFAOYSA-N beclobrate Chemical compound C1=CC(OC(C)(CC)C(=O)OCC)=CC=C1CC1=CC=C(Cl)C=C1 YWQGBCXVCXMSLJ-UHFFFAOYSA-N 0.000 description 1
- 229950009252 beclobrate Drugs 0.000 description 1
- 229950011170 beclotiamine Drugs 0.000 description 1
- 229950008861 befiperide Drugs 0.000 description 1
- ZPQPDBIHYCBNIG-UHFFFAOYSA-N befunolol Chemical compound CC(C)NCC(O)COC1=CC=CC2=C1OC(C(C)=O)=C2 ZPQPDBIHYCBNIG-UHFFFAOYSA-N 0.000 description 1
- 229960004374 befunolol Drugs 0.000 description 1
- SRIJFPBZWUFLFD-UHFFFAOYSA-N befuraline Chemical compound C=1C2=CC=CC=C2OC=1C(=O)N(CC1)CCN1CC1=CC=CC=C1 SRIJFPBZWUFLFD-UHFFFAOYSA-N 0.000 description 1
- 229950000159 befuraline Drugs 0.000 description 1
- 229960001192 bekanamycin Drugs 0.000 description 1
- 229950008989 belarizine Drugs 0.000 description 1
- NUTAQRYCLMZJIK-UHFFFAOYSA-N beloxamide Chemical compound C=1C=CC=CC=1CON(C(=O)C)CCCC1=CC=CC=C1 NUTAQRYCLMZJIK-UHFFFAOYSA-N 0.000 description 1
- 229950001610 beloxamide Drugs 0.000 description 1
- 229950003712 bemarinone Drugs 0.000 description 1
- 229960000750 bemegride Drugs 0.000 description 1
- PYVUMAGVCSQCBD-UHFFFAOYSA-N bemetizide Chemical compound N1C2=CC(Cl)=C(S(N)(=O)=O)C=C2S(=O)(=O)NC1C(C)C1=CC=CC=C1 PYVUMAGVCSQCBD-UHFFFAOYSA-N 0.000 description 1
- 229950004310 bemetizide Drugs 0.000 description 1
- 229950004294 bemitradine Drugs 0.000 description 1
- 229960001498 benactyzine Drugs 0.000 description 1
- 229950006837 benafentrine Drugs 0.000 description 1
- 229950005131 benaxibine Drugs 0.000 description 1
- 229960004530 benazepril Drugs 0.000 description 1
- 229950003634 bencianol Drugs 0.000 description 1
- 229950001938 bencisteine Drugs 0.000 description 1
- 229950000161 benclonidine Drugs 0.000 description 1
- 229960000945 bencyclane Drugs 0.000 description 1
- FYJJXENSONZJRG-UHFFFAOYSA-N bencyclane Chemical compound C=1C=CC=CC=1CC1(OCCCN(C)C)CCCCCC1 FYJJXENSONZJRG-UHFFFAOYSA-N 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- 229960005149 bendazac Drugs 0.000 description 1
- BYFMCKSPFYVMOU-UHFFFAOYSA-N bendazac Chemical compound C12=CC=CC=C2C(OCC(=O)O)=NN1CC1=CC=CC=C1 BYFMCKSPFYVMOU-UHFFFAOYSA-N 0.000 description 1
- YTLQFZVCLXFFRK-UHFFFAOYSA-N bendazol Chemical compound N=1C2=CC=CC=C2NC=1CC1=CC=CC=C1 YTLQFZVCLXFFRK-UHFFFAOYSA-N 0.000 description 1
- 229950000900 bendazol Drugs 0.000 description 1
- 229950004334 benderizine Drugs 0.000 description 1
- 229960003515 bendroflumethiazide Drugs 0.000 description 1
- HDWIHXWEUNVBIY-UHFFFAOYSA-N bendroflumethiazidum Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2CC1=CC=CC=C1 HDWIHXWEUNVBIY-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229950005162 benexate Drugs 0.000 description 1
- 229950008005 benfosformin Drugs 0.000 description 1
- 229960002873 benfotiamine Drugs 0.000 description 1
- BTNNPSLJPBRMLZ-LGMDPLHJSA-N benfotiamine Chemical compound C=1C=CC=CC=1C(=O)SC(/CCOP(O)(O)=O)=C(/C)N(C=O)CC1=CN=C(C)N=C1N BTNNPSLJPBRMLZ-LGMDPLHJSA-N 0.000 description 1
- 229950010443 benfurodil hemisuccinate Drugs 0.000 description 1
- 229950006196 benhepazone Drugs 0.000 description 1
- 229960004916 benidipine Drugs 0.000 description 1
- QZVNQOLPLYWLHQ-ZEQKJWHPSA-N benidipine Chemical compound C1([C@H]2C(=C(C)NC(C)=C2C(=O)OC)C(=O)O[C@H]2CN(CC=3C=CC=CC=3)CCC2)=CC=CC([N+]([O-])=O)=C1 QZVNQOLPLYWLHQ-ZEQKJWHPSA-N 0.000 description 1
- BEWNZPMDJIGBED-UHFFFAOYSA-N benmoxin Chemical compound C=1C=CC=CC=1C(C)NNC(=O)C1=CC=CC=C1 BEWNZPMDJIGBED-UHFFFAOYSA-N 0.000 description 1
- 229950011271 benmoxin Drugs 0.000 description 1
- 229950007729 benolizime Drugs 0.000 description 1
- FEJKLNWAOXSSNR-UHFFFAOYSA-N benorilate Chemical compound C1=CC(NC(=O)C)=CC=C1OC(=O)C1=CC=CC=C1OC(C)=O FEJKLNWAOXSSNR-UHFFFAOYSA-N 0.000 description 1
- 229960004277 benorilate Drugs 0.000 description 1
- RQETXCPBBLHUIB-UGCZWRCOSA-N benorterone Chemical compound C1C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)CC2 RQETXCPBBLHUIB-UGCZWRCOSA-N 0.000 description 1
- 229950007580 benorterone Drugs 0.000 description 1
- 229950006402 benoxafos Drugs 0.000 description 1
- 229960005430 benoxaprofen Drugs 0.000 description 1
- 229960002507 benperidol Drugs 0.000 description 1
- 229960001871 benproperine Drugs 0.000 description 1
- 229950004650 benrixate Drugs 0.000 description 1
- 229950003657 bensalan Drugs 0.000 description 1
- BNQDCRGUHNALGH-UHFFFAOYSA-N benserazide Chemical compound OCC(N)C(=O)NNCC1=CC=C(O)C(O)=C1O BNQDCRGUHNALGH-UHFFFAOYSA-N 0.000 description 1
- 229960000911 benserazide Drugs 0.000 description 1
- 229950005738 bensuldazic acid Drugs 0.000 description 1
- AIZFEOPQVZBNGH-UHFFFAOYSA-N bentazepam Chemical compound C1=2C=3CCCCC=3SC=2NC(=O)CN=C1C1=CC=CC=C1 AIZFEOPQVZBNGH-UHFFFAOYSA-N 0.000 description 1
- 229950001957 bentazepam Drugs 0.000 description 1
- 229950007784 bentemazole Drugs 0.000 description 1
- 229950006791 bentiamine Drugs 0.000 description 1
- 229950008752 bentipimine Drugs 0.000 description 1
- SPPTWHFVYKCNNK-FQEVSTJZSA-N bentiromide Chemical compound C1=CC(C(=O)O)=CC=C1NC(=O)[C@@H](NC(=O)C=1C=CC=CC=1)CC1=CC=C(O)C=C1 SPPTWHFVYKCNNK-FQEVSTJZSA-N 0.000 description 1
- 229960002515 bentiromide Drugs 0.000 description 1
- JFZGBMJPJZDNNT-UHFFFAOYSA-N benurestat Chemical compound ONC(=O)CNC(=O)C1=CC=C(Cl)C=C1 JFZGBMJPJZDNNT-UHFFFAOYSA-N 0.000 description 1
- 229950000421 benurestat Drugs 0.000 description 1
- 229940095076 benzaldehyde Drugs 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- GNZCRYWFWKFIDM-UHFFFAOYSA-N benzamidosalicylate Chemical compound C1=C(O)C(C(=O)O)=CC=C1NC(=O)C1=CC=CC=C1 GNZCRYWFWKFIDM-UHFFFAOYSA-N 0.000 description 1
- 229950002199 benzaprinoxide Drugs 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- 229960003545 benzarone Drugs 0.000 description 1
- 229960001081 benzatropine Drugs 0.000 description 1
- GIJXKZJWITVLHI-PMOLBWCYSA-N benzatropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(C=1C=CC=CC=1)C1=CC=CC=C1 GIJXKZJWITVLHI-PMOLBWCYSA-N 0.000 description 1
- 229960002529 benzbromarone Drugs 0.000 description 1
- WHQCHUCQKNIQEC-UHFFFAOYSA-N benzbromarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(Br)=C(O)C(Br)=C1 WHQCHUCQKNIQEC-UHFFFAOYSA-N 0.000 description 1
- DUTFBSAKKUNBAL-UHFFFAOYSA-N benzestrol Chemical compound C=1C=C(O)C=CC=1C(CC)C(CC)C(C)C1=CC=C(O)C=C1 DUTFBSAKKUNBAL-UHFFFAOYSA-N 0.000 description 1
- 229950000135 benzestrol Drugs 0.000 description 1
- UVTBZAWTRVBTMK-UHFFFAOYSA-N benzethidine Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCOCC1=CC=CC=C1 UVTBZAWTRVBTMK-UHFFFAOYSA-N 0.000 description 1
- 229950002302 benzethidine Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- 229950001299 benzetimide Drugs 0.000 description 1
- 229940065295 benzilone Drugs 0.000 description 1
- ZKCWITXZGWUJAV-UHFFFAOYSA-N benzilonium Chemical compound C1[N+](CC)(CC)CCC1OC(=O)C(O)(C=1C=CC=CC=1)C1=CC=CC=C1 ZKCWITXZGWUJAV-UHFFFAOYSA-N 0.000 description 1
- 229950000834 benzindopyrine Drugs 0.000 description 1
- 229960004411 benziodarone Drugs 0.000 description 1
- CZCHIEJNWPNBDE-UHFFFAOYSA-N benziodarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(O)C(I)=C1 CZCHIEJNWPNBDE-UHFFFAOYSA-N 0.000 description 1
- 229950000883 benzmalecene Drugs 0.000 description 1
- 229960004001 benznidazole Drugs 0.000 description 1
- QMOWPJIFTHVQMB-UHFFFAOYSA-N benzobarbital Chemical compound O=C1C(CC)(C=2C=CC=CC=2)C(=O)NC(=O)N1C(=O)C1=CC=CC=C1 QMOWPJIFTHVQMB-UHFFFAOYSA-N 0.000 description 1
- 229950010049 benzobarbital Drugs 0.000 description 1
- 229960005274 benzocaine Drugs 0.000 description 1
- 229950006476 benzoclidine Drugs 0.000 description 1
- 229950005567 benzodepa Drugs 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- JBIROUFYLSSYDX-UHFFFAOYSA-M benzododecinium chloride Chemical compound [Cl-].CCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 JBIROUFYLSSYDX-UHFFFAOYSA-M 0.000 description 1
- 229960004074 benzododecinium chloride Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- BHKPHCKISVSDGV-UHFFFAOYSA-N benzoic acid 8-quinolinyl ester Chemical compound C=1C=CC2=CC=CN=C2C=1OC(=O)C1=CC=CC=C1 BHKPHCKISVSDGV-UHFFFAOYSA-N 0.000 description 1
- 229960002130 benzoin Drugs 0.000 description 1
- 229960003789 benzonatate Drugs 0.000 description 1
- MAFMQEKGGFWBAB-UHFFFAOYSA-N benzonatate Chemical compound CCCCNC1=CC=C(C(=O)OCCOCCOCCOCCOCCOCCOCCOCCOCCOC)C=C1 MAFMQEKGGFWBAB-UHFFFAOYSA-N 0.000 description 1
- 229950000149 benzopyrronium bromide Drugs 0.000 description 1
- QONVRTOGBZRRQT-GMUIIQOCSA-N benzopyrronium bromide Chemical compound C(c1ccccc1)(c1ccccc1)(C(=O)O[C@@H]1CC[N+](C1)(C)C)O.[BrH-] QONVRTOGBZRRQT-GMUIIQOCSA-N 0.000 description 1
- 229950006062 benzotript Drugs 0.000 description 1
- 229960001279 benzoxiquine Drugs 0.000 description 1
- 229960001574 benzoxonium chloride Drugs 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- 229960003328 benzoyl peroxide Drugs 0.000 description 1
- YXKTVDFXDRQTKV-HNNXBMFYSA-N benzphetamine Chemical compound C([C@H](C)N(C)CC=1C=CC=CC=1)C1=CC=CC=C1 YXKTVDFXDRQTKV-HNNXBMFYSA-N 0.000 description 1
- 229960002837 benzphetamine Drugs 0.000 description 1
- KMGARVOVYXNAOF-UHFFFAOYSA-N benzpiperylone Chemical compound C1CN(C)CCC1N1C(=O)C(CC=2C=CC=CC=2)=C(C=2C=CC=CC=2)N1 KMGARVOVYXNAOF-UHFFFAOYSA-N 0.000 description 1
- 229950007647 benzpiperylone Drugs 0.000 description 1
- 229950010268 benzpyrinium bromide Drugs 0.000 description 1
- CSQNIJRRXIHHAY-UHFFFAOYSA-N benzquercin Chemical compound C=1C=C(OCC=2C=CC=CC=2)C(OCC=2C=CC=CC=2)=CC=1C=1OC2=CC(OCC=3C=CC=CC=3)=CC(OCC=3C=CC=CC=3)=C2C(=O)C=1OCC1=CC=CC=C1 CSQNIJRRXIHHAY-UHFFFAOYSA-N 0.000 description 1
- 229960004802 benzquercin Drugs 0.000 description 1
- 229960004564 benzquinamide Drugs 0.000 description 1
- NDTSRXAMMQDVSW-UHFFFAOYSA-N benzthiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1N=C2CSCC1=CC=CC=C1 NDTSRXAMMQDVSW-UHFFFAOYSA-N 0.000 description 1
- 229960001541 benzthiazide Drugs 0.000 description 1
- 229960000333 benzydamine Drugs 0.000 description 1
- UVOSLKVETODLTR-CXNSMIOJSA-N benzyl n-[(2r)-1-[2,4-dibromo-6-[[cyclohexyl(methyl)amino]methyl]anilino]-3-[[(2r)-3-[2,4-dibromo-6-[[cyclohexyl(methyl)amino]methyl]anilino]-3-oxo-2-(phenylmethoxycarbonylamino)propyl]disulfanyl]-1-oxopropan-2-yl]carbamate Chemical compound N([C@@H](CSSC[C@@H](C(=O)NC1=C(Br)C=C(Br)C=C1CN(C)C1CCCCC1)NC(=O)OCC=1C=CC=CC=1)C(=O)NC=1C(=CC(Br)=CC=1Br)CN(C)C1CCCCC1)C(=O)OCC1=CC=CC=C1 UVOSLKVETODLTR-CXNSMIOJSA-N 0.000 description 1
- VFIUCBTYGKMLCM-UHFFFAOYSA-N benzyl n-[bis(aziridin-1-yl)phosphoryl]carbamate Chemical compound C=1C=CC=CC=1COC(=O)NP(=O)(N1CC1)N1CC1 VFIUCBTYGKMLCM-UHFFFAOYSA-N 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 229950005348 benzylsulfamide Drugs 0.000 description 1
- HLIBJQGJVDHCNB-UHFFFAOYSA-N benzylsulfamide Chemical compound C1=CC(S(=O)(=O)N)=CC=C1NCC1=CC=CC=C1 HLIBJQGJVDHCNB-UHFFFAOYSA-N 0.000 description 1
- 229950000100 beperidium iodide Drugs 0.000 description 1
- AVWWVJUMXRXPNF-UHFFFAOYSA-N bephenium Chemical compound C=1C=CC=CC=1C[N+](C)(C)CCOC1=CC=CC=C1 AVWWVJUMXRXPNF-UHFFFAOYSA-N 0.000 description 1
- 229960000254 bephenium Drugs 0.000 description 1
- 229950010579 bepiastine Drugs 0.000 description 1
- 229960003665 bepridil Drugs 0.000 description 1
- UIEATEWHFDRYRU-UHFFFAOYSA-N bepridil Chemical compound C1CCCN1C(COCC(C)C)CN(C=1C=CC=CC=1)CC1=CC=CC=C1 UIEATEWHFDRYRU-UHFFFAOYSA-N 0.000 description 1
- 229960002890 beraprost Drugs 0.000 description 1
- CTPOHARTNNSRSR-APJZLKAGSA-N beraprost Chemical compound O([C@H]1C[C@@H](O)[C@@H]([C@@H]21)/C=C/[C@@H](O)C(C)CC#CC)C1=C2C=CC=C1CCCC(O)=O CTPOHARTNNSRSR-APJZLKAGSA-N 0.000 description 1
- OJVABJMSSDUECT-UHFFFAOYSA-L berberin sulfate Chemical compound [O-]S([O-])(=O)=O.C1=C2CC[N+]3=CC4=C(OC)C(OC)=CC=C4C=C3C2=CC2=C1OCO2.C1=C2CC[N+]3=CC4=C(OC)C(OC)=CC=C4C=C3C2=CC2=C1OCO2 OJVABJMSSDUECT-UHFFFAOYSA-L 0.000 description 1
- REHLODZXMGOGQP-UHFFFAOYSA-N bermoprofen Chemical compound C1C(=O)C2=CC(C(C(O)=O)C)=CC=C2OC2=CC=C(C)C=C21 REHLODZXMGOGQP-UHFFFAOYSA-N 0.000 description 1
- 229950007517 bermoprofen Drugs 0.000 description 1
- 229950002013 berythromycin Drugs 0.000 description 1
- 229950004182 besulpamide Drugs 0.000 description 1
- 108010023562 beta 2-Glycoprotein I Proteins 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- VOIFKEWOFUNPBN-QIUUJYRFSA-N beta-D-glucuronamide Chemical compound NC(=O)[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O VOIFKEWOFUNPBN-QIUUJYRFSA-N 0.000 description 1
- 235000013734 beta-carotene Nutrition 0.000 description 1
- 239000011648 beta-carotene Substances 0.000 description 1
- TUPZEYHYWIEDIH-WAIFQNFQSA-N beta-carotene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCCC1(C)C)C=CC=C(/C)C=CC2=CCCCC2(C)C TUPZEYHYWIEDIH-WAIFQNFQSA-N 0.000 description 1
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 1
- 235000011175 beta-cyclodextrine Nutrition 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 229960002747 betacarotene Drugs 0.000 description 1
- XBMIVRRWGCYBTQ-GCJKJVERSA-N betacetylmethadol Chemical compound C=1C=CC=CC=1C(C[C@@H](C)N(C)C)([C@@H](OC(C)=O)CC)C1=CC=CC=C1 XBMIVRRWGCYBTQ-GCJKJVERSA-N 0.000 description 1
- 229950003254 betacetylmethadol Drugs 0.000 description 1
- 229960004853 betadex Drugs 0.000 description 1
- 229960004536 betahistine Drugs 0.000 description 1
- UUQMNUMQCIQDMZ-UHFFFAOYSA-N betahistine Chemical compound CNCCC1=CC=CC=N1 UUQMNUMQCIQDMZ-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229950004879 betameprodine Drugs 0.000 description 1
- QIRAYNIFEOXSPW-XLIONFOSSA-N betamethadol Chemical compound C=1C=CC=CC=1C(C[C@@H](C)N(C)C)([C@@H](O)CC)C1=CC=CC=C1 QIRAYNIFEOXSPW-XLIONFOSSA-N 0.000 description 1
- 229950003767 betamethadol Drugs 0.000 description 1
- AKUJBENLRBOFTD-QZIXMDIESA-N betamethasone acetate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(C)=O)(O)[C@@]1(C)C[C@@H]2O AKUJBENLRBOFTD-QZIXMDIESA-N 0.000 description 1
- 229960004648 betamethasone acetate Drugs 0.000 description 1
- 229950003047 betamethasone acibutate Drugs 0.000 description 1
- KGNVBESWWSSQAN-QEVRMTOFSA-N betamethasone acibutate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(C)=O)(OC(=O)C(C)C)[C@@]1(C)C[C@@H]2O KGNVBESWWSSQAN-QEVRMTOFSA-N 0.000 description 1
- SOQJPQZCPBDOMF-YCUXZELOSA-N betamethasone benzoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(F)[C@@]4(C)C=CC(=O)C=C4CC[C@H]3[C@@H]2C[C@@H]1C)C(=O)CO)C(=O)C1=CC=CC=C1 SOQJPQZCPBDOMF-YCUXZELOSA-N 0.000 description 1
- 229960000870 betamethasone benzoate Drugs 0.000 description 1
- 229960001102 betamethasone dipropionate Drugs 0.000 description 1
- CIWBQSYVNNPZIQ-XYWKZLDCSA-N betamethasone dipropionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CIWBQSYVNNPZIQ-XYWKZLDCSA-N 0.000 description 1
- VQODGRNSFPNSQE-DVTGEIKXSA-N betamethasone phosphate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COP(O)(O)=O)(O)[C@@]1(C)C[C@@H]2O VQODGRNSFPNSQE-DVTGEIKXSA-N 0.000 description 1
- 229950006991 betamethasone phosphate Drugs 0.000 description 1
- SNHRLVCMMWUAJD-SUYDQAKGSA-N betamethasone valerate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(OC(=O)CCCC)[C@@]1(C)C[C@@H]2O SNHRLVCMMWUAJD-SUYDQAKGSA-N 0.000 description 1
- 229960004311 betamethasone valerate Drugs 0.000 description 1
- RHRAMPXHWHSKQB-GGEUKFTFSA-N betamicin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CN)O2)O)[C@@H](N)C[C@H]1N RHRAMPXHWHSKQB-GGEUKFTFSA-N 0.000 description 1
- 229950001961 betamicin Drugs 0.000 description 1
- 229960004980 betanidine Drugs 0.000 description 1
- 229950000011 betaprodine Drugs 0.000 description 1
- 229960004324 betaxolol Drugs 0.000 description 1
- NWIUTZDMDHAVTP-UHFFFAOYSA-N betaxolol Chemical compound C1=CC(OCC(O)CNC(C)C)=CC=C1CCOCC1CC1 NWIUTZDMDHAVTP-UHFFFAOYSA-N 0.000 description 1
- 229960002114 betazole Drugs 0.000 description 1
- LLUJWFHAQXWJOF-UHFFFAOYSA-N betazole Chemical compound NCCC1=CC=N[N]1 LLUJWFHAQXWJOF-UHFFFAOYSA-N 0.000 description 1
- XXRMYXBSBOVVBH-UHFFFAOYSA-N bethanechol chloride Chemical compound [Cl-].C[N+](C)(C)CC(C)OC(N)=O XXRMYXBSBOVVBH-UHFFFAOYSA-N 0.000 description 1
- 229960002123 bethanechol chloride Drugs 0.000 description 1
- NIVZHWNOUVJHKV-UHFFFAOYSA-N bethanidine Chemical compound CN\C(=N/C)NCC1=CC=CC=C1 NIVZHWNOUVJHKV-UHFFFAOYSA-N 0.000 description 1
- VDPYMEBVIDZKMD-UHFFFAOYSA-N betiatide Chemical compound OC(=O)CNC(=O)CNC(=O)CNC(=O)CSC(=O)C1=CC=CC=C1 VDPYMEBVIDZKMD-UHFFFAOYSA-N 0.000 description 1
- 229950002258 betiatide Drugs 0.000 description 1
- 229950005028 betoxycaine Drugs 0.000 description 1
- CXYOBRKOFHQONJ-UHFFFAOYSA-N betoxycaine Chemical compound CCCCOC1=CC=C(C(=O)OCCOCCN(CC)CC)C=C1N CXYOBRKOFHQONJ-UHFFFAOYSA-N 0.000 description 1
- HXLAFSUPPDYFEO-UHFFFAOYSA-N bevantolol Chemical compound C1=C(OC)C(OC)=CC=C1CCNCC(O)COC1=CC=CC(C)=C1 HXLAFSUPPDYFEO-UHFFFAOYSA-N 0.000 description 1
- 229960003588 bevantolol Drugs 0.000 description 1
- AXKJGGRSAVLXTE-UHFFFAOYSA-M bevonium methyl sulfate Chemical compound COS([O-])(=O)=O.C[N+]1(C)CCCCC1COC(=O)C(O)(C=1C=CC=CC=1)C1=CC=CC=C1 AXKJGGRSAVLXTE-UHFFFAOYSA-M 0.000 description 1
- 229950000445 bevonium metilsulfate Drugs 0.000 description 1
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 1
- 229960000516 bezafibrate Drugs 0.000 description 1
- 229960004611 bezitramide Drugs 0.000 description 1
- FLKWNFFCSSJANB-UHFFFAOYSA-N bezitramide Chemical compound O=C1N(C(=O)CC)C2=CC=CC=C2N1C(CC1)CCN1CCC(C#N)(C=1C=CC=CC=1)C1=CC=CC=C1 FLKWNFFCSSJANB-UHFFFAOYSA-N 0.000 description 1
- DQNIWUUHJSXGHW-UHFFFAOYSA-N bialamicol Chemical compound C=CCC1=C(O)C(CN(CC)CC)=CC(C=2C=C(CN(CC)CC)C(O)=C(CC=C)C=2)=C1 DQNIWUUHJSXGHW-UHFFFAOYSA-N 0.000 description 1
- 229950000260 bialamicol Drugs 0.000 description 1
- 229960005140 bibenzonium bromide Drugs 0.000 description 1
- APVMLVNTOWQOHL-UHFFFAOYSA-M bibenzonium bromide Chemical compound [Br-].C=1C=CC=CC=1C(OCC[N+](C)(C)C)CC1=CC=CC=C1 APVMLVNTOWQOHL-UHFFFAOYSA-M 0.000 description 1
- 229960002008 bibrocathol Drugs 0.000 description 1
- VTAVFIZOZUAKKE-UHFFFAOYSA-K bibrocathol Chemical compound BrC1=C(Br)C(Br)=C(Br)C2=C1O[Bi](O)O2 VTAVFIZOZUAKKE-UHFFFAOYSA-K 0.000 description 1
- 229950010365 bicifadine Drugs 0.000 description 1
- OFYVIGTWSQPCLF-NWDGAFQWSA-N bicifadine Chemical compound C1=CC(C)=CC=C1[C@@]1(CNC2)[C@H]2C1 OFYVIGTWSQPCLF-NWDGAFQWSA-N 0.000 description 1
- 229950006778 biclodil Drugs 0.000 description 1
- 229950002800 biclofibrate Drugs 0.000 description 1
- 229960004093 biclotymol Drugs 0.000 description 1
- HNOOXWDWUSLXOB-UHFFFAOYSA-N biclotymol Chemical compound CC(C)C1=CC(Cl)=C(C)C(CC=2C(=C(C(C)C)C=C(Cl)C=2C)O)=C1O HNOOXWDWUSLXOB-UHFFFAOYSA-N 0.000 description 1
- WOUDXEYYJPOSNE-VKZDFBPFSA-N bicozamycin Chemical compound N1C(=O)[C@@]2(O)NC(=O)[C@]1([C@@H](O)[C@@](O)(CO)C)OCCC2=C WOUDXEYYJPOSNE-VKZDFBPFSA-N 0.000 description 1
- 229950007558 bicozamycin Drugs 0.000 description 1
- WOUDXEYYJPOSNE-UHFFFAOYSA-N bicyclomycin Natural products N1C(=O)C2(O)NC(=O)C1(C(O)C(O)(CO)C)OCCC2=C WOUDXEYYJPOSNE-UHFFFAOYSA-N 0.000 description 1
- 229950005940 bietamiverine Drugs 0.000 description 1
- JGTJANXYSNVLMQ-UHFFFAOYSA-N bietamiverine Chemical compound C=1C=CC=CC=1C(C(=O)OCCN(CC)CC)N1CCCCC1 JGTJANXYSNVLMQ-UHFFFAOYSA-N 0.000 description 1
- 229960004383 bietaserpine Drugs 0.000 description 1
- 229960004933 bifemelane Drugs 0.000 description 1
- 229950004144 bifepramide Drugs 0.000 description 1
- 229950004654 bifluranol Drugs 0.000 description 1
- 229960002206 bifonazole Drugs 0.000 description 1
- 229950004874 binedaline Drugs 0.000 description 1
- SXYFFMXPDDGOEK-UHFFFAOYSA-N binedaline Chemical compound C12=CC=CC=C2N(N(C)CCN(C)C)C=C1C1=CC=CC=C1 SXYFFMXPDDGOEK-UHFFFAOYSA-N 0.000 description 1
- 229950011614 binfloxacin Drugs 0.000 description 1
- 229960001901 bioallethrin Drugs 0.000 description 1
- 229920000229 biodegradable polyester Polymers 0.000 description 1
- 239000004622 biodegradable polyester Substances 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 229950002373 bioresmethrin Drugs 0.000 description 1
- VEMKTZHHVJILDY-UXHICEINSA-N bioresmethrin Chemical compound CC1(C)[C@H](C=C(C)C)[C@H]1C(=O)OCC1=COC(CC=2C=CC=CC=2)=C1 VEMKTZHHVJILDY-UXHICEINSA-N 0.000 description 1
- 230000006287 biotinylation Effects 0.000 description 1
- 238000007413 biotinylation Methods 0.000 description 1
- 229950009454 bipenamol Drugs 0.000 description 1
- YSXKPIUOCJLQIE-UHFFFAOYSA-N biperiden Chemical compound C1C(C=C2)CC2C1C(C=1C=CC=CC=1)(O)CCN1CCCCC1 YSXKPIUOCJLQIE-UHFFFAOYSA-N 0.000 description 1
- 229960003003 biperiden Drugs 0.000 description 1
- QRZAKQDHEVVFRX-UHFFFAOYSA-N biphenyl-4-ylacetic acid Chemical compound C1=CC(CC(=O)O)=CC=C1C1=CC=CC=C1 QRZAKQDHEVVFRX-UHFFFAOYSA-N 0.000 description 1
- YCNCIZWAGQTWBI-UHFFFAOYSA-N biriperone Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CC2CC(C3=CC=CC=C3N3)=C3CN2CC1 YCNCIZWAGQTWBI-UHFFFAOYSA-N 0.000 description 1
- 229950001988 biriperone Drugs 0.000 description 1
- IEPBPSSCIZTJIF-UHFFFAOYSA-N bis(2,2,2-trichloroethyl) carbonate Chemical compound ClC(Cl)(Cl)COC(=O)OCC(Cl)(Cl)Cl IEPBPSSCIZTJIF-UHFFFAOYSA-N 0.000 description 1
- VWMVVFFLQCUGKR-UHFFFAOYSA-M bis(2,4-dichlorophenyl)iodanium;chloride Chemical compound [Cl-].ClC1=CC(Cl)=CC=C1[I+]C1=CC=C(Cl)C=C1Cl VWMVVFFLQCUGKR-UHFFFAOYSA-M 0.000 description 1
- YGCONRRHHMKQRL-UHFFFAOYSA-N bis(2-anilino-2-oxoethyl)-dimethylazanium;chloride Chemical compound [Cl-].C=1C=CC=CC=1NC(=O)C[N+](C)(C)CC(=O)NC1=CC=CC=C1 YGCONRRHHMKQRL-UHFFFAOYSA-N 0.000 description 1
- QKSKPIVNLNLAAV-UHFFFAOYSA-N bis(2-chloroethyl) sulfide Chemical compound ClCCSCCCl QKSKPIVNLNLAAV-UHFFFAOYSA-N 0.000 description 1
- 229960000503 bisacodyl Drugs 0.000 description 1
- 229950008548 bisantrene Drugs 0.000 description 1
- 229950004793 bisbendazole Drugs 0.000 description 1
- 229950009892 bisbentiamine Drugs 0.000 description 1
- 229950003394 bisfenazone Drugs 0.000 description 1
- 229950002342 bisfentidine Drugs 0.000 description 1
- JAONZGLTYYUPCT-UHFFFAOYSA-K bismuth subgallate Chemical compound OC(=O)C1=CC(O)=C2O[Bi](O)OC2=C1 JAONZGLTYYUPCT-UHFFFAOYSA-K 0.000 description 1
- 229960000199 bismuth subgallate Drugs 0.000 description 1
- ZREIPSZUJIFJNP-UHFFFAOYSA-K bismuth subsalicylate Chemical compound C1=CC=C2O[Bi](O)OC(=O)C2=C1 ZREIPSZUJIFJNP-UHFFFAOYSA-K 0.000 description 1
- 229960000782 bismuth subsalicylate Drugs 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- XUYANFPPYJSBPU-QMMMGPOBSA-N bisorcic Chemical compound CC(=O)NCCC[C@@H](C(O)=O)NC(C)=O XUYANFPPYJSBPU-QMMMGPOBSA-N 0.000 description 1
- 229950008082 bisorcic Drugs 0.000 description 1
- 229960002326 bithionol Drugs 0.000 description 1
- JFIOVJDNOJYLKP-UHFFFAOYSA-N bithionol Chemical compound OC1=C(Cl)C=C(Cl)C=C1SC1=CC(Cl)=CC(Cl)=C1O JFIOVJDNOJYLKP-UHFFFAOYSA-N 0.000 description 1
- 229950008258 bithionoloxide Drugs 0.000 description 1
- 229950006860 bitipazone Drugs 0.000 description 1
- 229960001500 bivalirudin Drugs 0.000 description 1
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 description 1
- 108010055460 bivalirudin Proteins 0.000 description 1
- 230000008081 blood perfusion Effects 0.000 description 1
- 229950004374 bluensomycin Drugs 0.000 description 1
- 229950003042 bofumustine Drugs 0.000 description 1
- JFAXVZXNIGIDDA-KDWXAGHCSA-N bolandiol dipropionate Chemical compound C1CC2=C[C@@H](OC(=O)CC)CC[C@@H]2[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CC)[C@@]1(C)CC2 JFAXVZXNIGIDDA-KDWXAGHCSA-N 0.000 description 1
- 229950004556 bolandiol dipropionate Drugs 0.000 description 1
- 229950008036 bolasterone Drugs 0.000 description 1
- BQDZMZRUXNFTQT-OHMLUKIUSA-N bolazine Chemical compound C([C@]1(C)[C@@H](O)CC[C@H]1[C@@H]1CC2)C[C@@H]1[C@](C[C@H]1C)(C)[C@@H]2C\C1=N/N=C1/C[C@H](CC[C@@H]2[C@@H]3CC[C@]4(C)[C@@H](O)CC[C@H]42)[C@]3(C)C[C@H]1C BQDZMZRUXNFTQT-OHMLUKIUSA-N 0.000 description 1
- 229950009014 bolazine Drugs 0.000 description 1
- VGXLQFPZGUQVQW-XGXHKTLJSA-N bolenol Chemical compound C1C=C2CCCC[C@@H]2[C@@H]2[C@@H]1[C@@H]1CC[C@](CC)(O)[C@@]1(C)CC2 VGXLQFPZGUQVQW-XGXHKTLJSA-N 0.000 description 1
- 229950007251 bolenol Drugs 0.000 description 1
- NCXAMLZZPQRJGY-PVAKYVEVSA-N bolmantalate Chemical compound C1C(C2)CC(C3)CC2CC13C(=O)O[C@H]1CC[C@H]2[C@H](CCC=3[C@@H]4CCC(=O)C=3)[C@@H]4CC[C@@]21C NCXAMLZZPQRJGY-PVAKYVEVSA-N 0.000 description 1
- 229950001400 bolmantalate Drugs 0.000 description 1
- 229950000729 bometolol Drugs 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 229960001035 bopindolol Drugs 0.000 description 1
- 229960001793 bornaprine Drugs 0.000 description 1
- 229950002803 bornaprolol Drugs 0.000 description 1
- 229950000757 bornelone Drugs 0.000 description 1
- 229950011443 botiacrine Drugs 0.000 description 1
- FOCUKKUAGZPPCU-UHFFFAOYSA-N boxidine Chemical compound C1=CC(C(F)(F)F)=CC=C1C(C=C1)=CC=C1OCCN1CCCC1 FOCUKKUAGZPPCU-UHFFFAOYSA-N 0.000 description 1
- 229950010104 boxidine Drugs 0.000 description 1
- QXZGBUJJYSLZLT-FDISYFBBSA-N bradykinin Chemical compound NC(=N)NCCC[C@H](N)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CO)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)CCC1 QXZGBUJJYSLZLT-FDISYFBBSA-N 0.000 description 1
- DYODAJAEQDVYFX-UHFFFAOYSA-N brallobarbital Chemical compound BrC(=C)CC1(CC=C)C(=O)NC(=O)NC1=O DYODAJAEQDVYFX-UHFFFAOYSA-N 0.000 description 1
- 229950002261 brallobarbital Drugs 0.000 description 1
- 229950010062 brazergoline Drugs 0.000 description 1
- YYUZPBGIBVJYSI-UHFFFAOYSA-N brefonalol Chemical compound C=1C=C2NC(=O)CCC2=CC=1C(O)CNC(C)(C)CCC1=CC=CC=C1 YYUZPBGIBVJYSI-UHFFFAOYSA-N 0.000 description 1
- 229950005827 brefonalol Drugs 0.000 description 1
- ZDXGFIXMPOUDFF-XLIONFOSSA-N bremazocine Chemical compound C([C@]1(C2=CC(O)=CC=C2C[C@@H]2C1(C)C)CC)CN2CC1(O)CC1 ZDXGFIXMPOUDFF-XLIONFOSSA-N 0.000 description 1
- 229950008841 bremazocine Drugs 0.000 description 1
- PHEZJEYUWHETKO-UHFFFAOYSA-N brequinar Chemical compound N1=C2C=CC(F)=CC2=C(C(O)=O)C(C)=C1C(C=C1)=CC=C1C1=CC=CC=C1F PHEZJEYUWHETKO-UHFFFAOYSA-N 0.000 description 1
- 229950010231 brequinar Drugs 0.000 description 1
- 229960004895 bretylium tosylate Drugs 0.000 description 1
- KVWNWTZZBKCOPM-UHFFFAOYSA-M bretylium tosylate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1.CC[N+](C)(C)CC1=CC=CC=C1Br KVWNWTZZBKCOPM-UHFFFAOYSA-M 0.000 description 1
- KIGNRDGJGXIHCH-DQUMZJRKSA-N brindoxime Chemical compound C([C@@H](C)O\N=C1/c2c(cc(cc2c2c1ncnc2)Br)Br)(=O)NCCN(C)C KIGNRDGJGXIHCH-DQUMZJRKSA-N 0.000 description 1
- 229950007589 brindoxime Drugs 0.000 description 1
- 229950007491 brobactam Drugs 0.000 description 1
- 229950004691 broclepride Drugs 0.000 description 1
- 229950000394 brocresine Drugs 0.000 description 1
- 229950002584 brocrinat Drugs 0.000 description 1
- BFCRRLMMHNLSCP-UHFFFAOYSA-N brodimoprim Chemical compound COC1=C(Br)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 BFCRRLMMHNLSCP-UHFFFAOYSA-N 0.000 description 1
- 229960000252 brodimoprim Drugs 0.000 description 1
- WZXHSWVDAYOFPE-UHFFFAOYSA-N brofaromine Chemical compound C=1C2=CC(OC)=CC(Br)=C2OC=1C1CCNCC1 WZXHSWVDAYOFPE-UHFFFAOYSA-N 0.000 description 1
- 229950004068 brofaromine Drugs 0.000 description 1
- 229950000656 brofezil Drugs 0.000 description 1
- 229950011563 brofoxine Drugs 0.000 description 1
- 229950010276 brolaconazole Drugs 0.000 description 1
- 229950009957 brolamfetamine Drugs 0.000 description 1
- 229950000114 bromacrylide Drugs 0.000 description 1
- UFDJFJYMMIZKLG-KBPBESRZSA-N bromadoline Chemical compound CN(C)[C@H]1CCCC[C@@H]1NC(=O)C1=CC=C(Br)C=C1 UFDJFJYMMIZKLG-KBPBESRZSA-N 0.000 description 1
- 229950005097 bromadoline Drugs 0.000 description 1
- 229960002729 bromazepam Drugs 0.000 description 1
- 229960003166 bromazine Drugs 0.000 description 1
- NUNIWXHYABYXKF-UHFFFAOYSA-N bromazine Chemical compound C=1C=C(Br)C=CC=1C(OCCN(C)C)C1=CC=CC=C1 NUNIWXHYABYXKF-UHFFFAOYSA-N 0.000 description 1
- 229950002954 bromchlorenone Drugs 0.000 description 1
- UPZFHUODAYGHDZ-RMKNXTFCSA-N bromebric acid Chemical compound COC1=CC=C(C(=O)C(\Br)=C/C(O)=O)C=C1 UPZFHUODAYGHDZ-RMKNXTFCSA-N 0.000 description 1
- 229950009886 bromebric acid Drugs 0.000 description 1
- 229950006651 bromerguride Drugs 0.000 description 1
- OXBIWQBTTCRDGT-UHFFFAOYSA-N brometenamine Chemical compound BrC(Br)Br.C1N(C2)CN3CN1CN2C3 OXBIWQBTTCRDGT-UHFFFAOYSA-N 0.000 description 1
- 229950008799 brometenamine Drugs 0.000 description 1
- 229960003655 bromfenac Drugs 0.000 description 1
- ZBPLOVFIXSTCRZ-UHFFFAOYSA-N bromfenac Chemical compound NC1=C(CC(O)=O)C=CC=C1C(=O)C1=CC=C(Br)C=C1 ZBPLOVFIXSTCRZ-UHFFFAOYSA-N 0.000 description 1
- 229960003870 bromhexine Drugs 0.000 description 1
- OJGDCBLYJGHCIH-UHFFFAOYSA-N bromhexine Chemical compound C1CCCCC1N(C)CC1=CC(Br)=CC(Br)=C1N OJGDCBLYJGHCIH-UHFFFAOYSA-N 0.000 description 1
- 229950004502 bromindione Drugs 0.000 description 1
- NPUZIGSOEWMFKK-UHFFFAOYSA-N bromindione Chemical compound C1=CC(Br)=CC=C1C1C(=O)C2=CC=CC=C2C1=O NPUZIGSOEWMFKK-UHFFFAOYSA-N 0.000 description 1
- 229960003880 bromisoval Drugs 0.000 description 1
- MEXUFEQDCXZEON-UHFFFAOYSA-N bromochlorodifluoromethane Chemical compound FC(F)(Cl)Br MEXUFEQDCXZEON-UHFFFAOYSA-N 0.000 description 1
- 229950000861 bromociclen Drugs 0.000 description 1
- 229960002802 bromocriptine Drugs 0.000 description 1
- OZVBMTJYIDMWIL-AYFBDAFISA-N bromocriptine Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 OZVBMTJYIDMWIL-AYFBDAFISA-N 0.000 description 1
- 229950006751 bromofenofos Drugs 0.000 description 1
- 229960001034 bromopride Drugs 0.000 description 1
- RJCQBQGAPKAMLL-UHFFFAOYSA-N bromotrifluoromethane Chemical compound FC(F)(F)Br RJCQBQGAPKAMLL-UHFFFAOYSA-N 0.000 description 1
- 229960004037 bromperidol Drugs 0.000 description 1
- 229960004284 bromperidol decanoate Drugs 0.000 description 1
- ZDIGNSYAACHWNL-UHFFFAOYSA-N brompheniramine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Br)C=C1 ZDIGNSYAACHWNL-UHFFFAOYSA-N 0.000 description 1
- 229960000725 brompheniramine Drugs 0.000 description 1
- 229960003168 bronopol Drugs 0.000 description 1
- 229950003738 broparestrol Drugs 0.000 description 1
- 229950011622 broperamole Drugs 0.000 description 1
- 229950009494 bropirimine Drugs 0.000 description 1
- 229950010711 broquinaldol Drugs 0.000 description 1
- 229950001568 brosotamide Drugs 0.000 description 1
- 229950006280 brosuximide Drugs 0.000 description 1
- 229950005372 brotianide Drugs 0.000 description 1
- 229960003051 brotizolam Drugs 0.000 description 1
- DQTRREPKGJIABH-UHFFFAOYSA-N brovanexine Chemical compound C1=C(OC(C)=O)C(OC)=CC(C(=O)NC=2C(=CC(Br)=CC=2Br)CN(C)C2CCCCC2)=C1 DQTRREPKGJIABH-UHFFFAOYSA-N 0.000 description 1
- 229960001835 brovanexine Drugs 0.000 description 1
- WYIJGAVIVKPUGJ-GIVPXCGWSA-N brovincamine Chemical compound BrC1=CC=C2C(CCN3CCC4)=C5[C@@H]3[C@]4(CC)C[C@](O)(C(=O)OC)N5C2=C1 WYIJGAVIVKPUGJ-GIVPXCGWSA-N 0.000 description 1
- 229950002641 brovincamine Drugs 0.000 description 1
- 229950002992 broxaldine Drugs 0.000 description 1
- JBRBWHCVRGURBA-UHFFFAOYSA-N broxaterol Chemical compound CC(C)(C)NCC(O)C1=CC(Br)=NO1 JBRBWHCVRGURBA-UHFFFAOYSA-N 0.000 description 1
- 229950008847 broxaterol Drugs 0.000 description 1
- 229950003335 broxitalamic acid Drugs 0.000 description 1
- 229950004398 broxuridine Drugs 0.000 description 1
- 229960002880 broxyquinoline Drugs 0.000 description 1
- IRQXZTBHNKVIRL-AYXPYFKUSA-N bruceantin Natural products CC1=C(O)C(=O)C[C@]2(C)[C@@H]([C@@H](O)[C@@H]3O)[C@@]45CO[C@@]3(C(=O)OC)[C@@H]5[C@@H](OC(=O)C=C(C)C(C)C)C(=O)O[C@@H]4C[C@H]21 IRQXZTBHNKVIRL-AYXPYFKUSA-N 0.000 description 1
- RRKTZKIUPZVBMF-IBTVXLQLSA-N brucine Chemical compound O([C@@H]1[C@H]([C@H]2C3)[C@@H]4N(C(C1)=O)C=1C=C(C(=CC=11)OC)OC)CC=C2CN2[C@@H]3[C@]41CC2 RRKTZKIUPZVBMF-IBTVXLQLSA-N 0.000 description 1
- RRKTZKIUPZVBMF-UHFFFAOYSA-N brucine Natural products C1=2C=C(OC)C(OC)=CC=2N(C(C2)=O)C3C(C4C5)C2OCC=C4CN2C5C31CC2 RRKTZKIUPZVBMF-UHFFFAOYSA-N 0.000 description 1
- 229950003382 bucainide Drugs 0.000 description 1
- 229960005470 bucetin Drugs 0.000 description 1
- 229950003665 buciclovir Drugs 0.000 description 1
- 229960004272 bucillamine Drugs 0.000 description 1
- 229950005341 bucindolol Drugs 0.000 description 1
- CJGYSWNGNKCJSB-YVLZZHOMSA-N bucladesine Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](OC(=O)CCC)[C@@H]2N1C(N=CN=C2NC(=O)CCC)=C2N=C1 CJGYSWNGNKCJSB-YVLZZHOMSA-N 0.000 description 1
- 229960005263 bucladesine Drugs 0.000 description 1
- MOYGZHXDRJNJEP-UHFFFAOYSA-N buclizine Chemical compound C1=CC(C(C)(C)C)=CC=C1CN1CCN(C(C=2C=CC=CC=2)C=2C=CC(Cl)=CC=2)CC1 MOYGZHXDRJNJEP-UHFFFAOYSA-N 0.000 description 1
- 229960001705 buclizine Drugs 0.000 description 1
- ZGJHIFYEQJEUKA-UHFFFAOYSA-N buclosamide Chemical compound CCCCNC(=O)C1=CC=C(Cl)C=C1O ZGJHIFYEQJEUKA-UHFFFAOYSA-N 0.000 description 1
- 229950000430 buclosamide Drugs 0.000 description 1
- IJTPQQVCKPZIMV-UHFFFAOYSA-N bucloxic acid Chemical compound ClC1=CC(C(=O)CCC(=O)O)=CC=C1C1CCCCC1 IJTPQQVCKPZIMV-UHFFFAOYSA-N 0.000 description 1
- 229950005608 bucloxic acid Drugs 0.000 description 1
- 229950003872 bucolome Drugs 0.000 description 1
- ZFPNOTAGQWNERF-UHFFFAOYSA-N bucricaine Chemical compound C1=CC=C2C(NCCCC)=C(CCCC3)C3=NC2=C1 ZFPNOTAGQWNERF-UHFFFAOYSA-N 0.000 description 1
- 229950006909 bucricaine Drugs 0.000 description 1
- DYGLNTZLBQBOPT-UHFFFAOYSA-N bucromarone Chemical compound C1=C(C)C(OCCCN(CCCC)CCCC)=C(C)C=C1C(=O)C1=CC(=O)C2=CC=CC=C2O1 DYGLNTZLBQBOPT-UHFFFAOYSA-N 0.000 description 1
- 229950005745 bucromarone Drugs 0.000 description 1
- CIJVBYRUFLGDHY-UHFFFAOYSA-N bucumolol Chemical compound O1C(=O)C=CC2=C1C(OCC(O)CNC(C)(C)C)=CC=C2C CIJVBYRUFLGDHY-UHFFFAOYSA-N 0.000 description 1
- 229950002568 bucumolol Drugs 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- QIHLUZAFSSMXHQ-UHFFFAOYSA-N budipine Chemical compound C1CN(C(C)(C)C)CCC1(C=1C=CC=CC=1)C1=CC=CC=C1 QIHLUZAFSSMXHQ-UHFFFAOYSA-N 0.000 description 1
- 229950002361 budotitane Drugs 0.000 description 1
- 229950001730 budralazine Drugs 0.000 description 1
- DQGFCLJXYFXXIJ-LFIBNONCSA-N budralazine Chemical compound C1=CC=C2C(N/N=C(C)/C=C(C)C)=NN=CC2=C1 DQGFCLJXYFXXIJ-LFIBNONCSA-N 0.000 description 1
- 229950006303 bufenadrine Drugs 0.000 description 1
- RFIXURDMUINBMD-UHFFFAOYSA-N bufeniode Chemical compound C=1C(I)=C(O)C(I)=CC=1C(O)C(C)NC(C)CCC1=CC=CC=C1 RFIXURDMUINBMD-UHFFFAOYSA-N 0.000 description 1
- 229950003250 bufeniode Drugs 0.000 description 1
- AKLNLVOZXMQGSI-UHFFFAOYSA-N bufetolol Chemical compound CC(C)(C)NCC(O)COC1=CC=CC=C1OCC1OCCC1 AKLNLVOZXMQGSI-UHFFFAOYSA-N 0.000 description 1
- 229950009385 bufetolol Drugs 0.000 description 1
- MXJWRABVEGLYDG-UHFFFAOYSA-N bufexamac Chemical compound CCCCOC1=CC=C(CC(=O)NO)C=C1 MXJWRABVEGLYDG-UHFFFAOYSA-N 0.000 description 1
- 229960000962 bufexamac Drugs 0.000 description 1
- 229950009075 bufezolac Drugs 0.000 description 1
- 229960001415 buflomedil Drugs 0.000 description 1
- 229950006858 bufogenin Drugs 0.000 description 1
- ATLJNLYIJOCWJE-CWMZOUAVSA-N bufogenin Chemical compound C=1([C@H]2C[C@H]3O[C@@]43[C@H]3[C@@H]([C@]5(CC[C@H](O)C[C@H]5CC3)C)CC[C@@]42C)C=CC(=O)OC=1 ATLJNLYIJOCWJE-CWMZOUAVSA-N 0.000 description 1
- 229960004111 buformin Drugs 0.000 description 1
- XSEUMFJMFFMCIU-UHFFFAOYSA-N buformin Chemical compound CCCC\N=C(/N)N=C(N)N XSEUMFJMFFMCIU-UHFFFAOYSA-N 0.000 description 1
- 229950001468 bufrolin Drugs 0.000 description 1
- 229950006886 bufuralol Drugs 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- FLWFHHFTIRLFPV-UHFFFAOYSA-N bumadizone Chemical compound C=1C=CC=CC=1N(C(=O)C(C(O)=O)CCCC)NC1=CC=CC=C1 FLWFHHFTIRLFPV-UHFFFAOYSA-N 0.000 description 1
- 229960003354 bumadizone Drugs 0.000 description 1
- 229950010833 bumecaine Drugs 0.000 description 1
- NJJPSFAJAKVSST-UHFFFAOYSA-N bumepidil Chemical compound CC1=NC2=NC=NN2C2=C1CCN2C(C)(C)C NJJPSFAJAKVSST-UHFFFAOYSA-N 0.000 description 1
- 229950004676 bumepidil Drugs 0.000 description 1
- 229960004064 bumetanide Drugs 0.000 description 1
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 1
- OCWYEMOEOGEQAN-UHFFFAOYSA-N bumetrizole Chemical compound CC(C)(C)C1=CC(C)=CC(N2N=C3C=C(Cl)C=CC3=N2)=C1O OCWYEMOEOGEQAN-UHFFFAOYSA-N 0.000 description 1
- 229950007917 bumetrizole Drugs 0.000 description 1
- 229960005458 bunaftine Drugs 0.000 description 1
- WWGZXRYELYWJBD-UHFFFAOYSA-N bunaftine Chemical compound C1=CC=C2C(C(=O)N(CCN(CC)CC)CCCC)=CC=CC2=C1 WWGZXRYELYWJBD-UHFFFAOYSA-N 0.000 description 1
- 229950004965 bunamidine Drugs 0.000 description 1
- CWRBDUIQDIHWJZ-SOFGYWHQSA-N bunamiodyl Chemical compound CCCC(=O)NC1=C(I)C=C(I)C(\C=C(/CC)C(O)=O)=C1I CWRBDUIQDIHWJZ-SOFGYWHQSA-N 0.000 description 1
- 229950006842 bunamiodyl Drugs 0.000 description 1
- 229950006427 bunaprolast Drugs 0.000 description 1
- 229960002467 bunazosin Drugs 0.000 description 1
- VCVQSRCYSKKPBA-UHFFFAOYSA-N bunitrolol Chemical compound CC(C)(C)NCC(O)COC1=CC=CC=C1C#N VCVQSRCYSKKPBA-UHFFFAOYSA-N 0.000 description 1
- 229950008581 bunitrolol Drugs 0.000 description 1
- 229950004443 bunolol Drugs 0.000 description 1
- 229950008349 buparvaquone Drugs 0.000 description 1
- 229950008162 bupicomide Drugs 0.000 description 1
- HQIRNZOQPUAHHV-UHFFFAOYSA-N bupranolol Chemical compound CC1=CC=C(Cl)C(OCC(O)CNC(C)(C)C)=C1 HQIRNZOQPUAHHV-UHFFFAOYSA-N 0.000 description 1
- 229960000330 bupranolol Drugs 0.000 description 1
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 description 1
- 229960001736 buprenorphine Drugs 0.000 description 1
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 description 1
- 229960001058 bupropion Drugs 0.000 description 1
- 229950000759 buquineran Drugs 0.000 description 1
- 229950003639 buquinolate Drugs 0.000 description 1
- 229950011280 buquiterine Drugs 0.000 description 1
- YFLRYAVDPKONNX-UHFFFAOYSA-N buramate Chemical compound OCCOC(=O)NCC1=CC=CC=C1 YFLRYAVDPKONNX-UHFFFAOYSA-N 0.000 description 1
- 229950009824 buramate Drugs 0.000 description 1
- 229950008929 burodiline Drugs 0.000 description 1
- 230000009172 bursting Effects 0.000 description 1
- QWCRAEMEVRGPNT-UHFFFAOYSA-N buspirone Chemical compound C1C(=O)N(CCCCN2CCN(CC2)C=2N=CC=CN=2)C(=O)CC21CCCC2 QWCRAEMEVRGPNT-UHFFFAOYSA-N 0.000 description 1
- 229960002495 buspirone Drugs 0.000 description 1
- 229940015694 butabarbital Drugs 0.000 description 1
- 229940068366 butabarbital sodium Drugs 0.000 description 1
- 229960003369 butacaine Drugs 0.000 description 1
- QTNZYVAMNRDUAD-UHFFFAOYSA-N butacetin Chemical compound CC(=O)NC1=CC=C(OC(C)(C)C)C=C1 QTNZYVAMNRDUAD-UHFFFAOYSA-N 0.000 description 1
- 229950011189 butacetin Drugs 0.000 description 1
- 229950006479 butaclamol Drugs 0.000 description 1
- 229950001848 butadiazamide Drugs 0.000 description 1
- 229960002134 butafosfan Drugs 0.000 description 1
- VYWQZAARVNRSTR-UHFFFAOYSA-N butalamine Chemical compound O1C(NCCN(CCCC)CCCC)=NC(C=2C=CC=CC=2)=N1 VYWQZAARVNRSTR-UHFFFAOYSA-N 0.000 description 1
- 229960003756 butalamine Drugs 0.000 description 1
- UZVHFVZFNXBMQJ-UHFFFAOYSA-N butalbital Chemical compound CC(C)CC1(CC=C)C(=O)NC(=O)NC1=O UZVHFVZFNXBMQJ-UHFFFAOYSA-N 0.000 description 1
- 229960002546 butalbital Drugs 0.000 description 1
- IUWVALYLNVXWKX-UHFFFAOYSA-N butamben Chemical compound CCCCOC(=O)C1=CC=C(N)C=C1 IUWVALYLNVXWKX-UHFFFAOYSA-N 0.000 description 1
- 229960000400 butamben Drugs 0.000 description 1
- 229960004902 butamirate Drugs 0.000 description 1
- DDVUMDPCZWBYRA-UHFFFAOYSA-N butamirate Chemical compound CCN(CC)CCOCCOC(=O)C(CC)C1=CC=CC=C1 DDVUMDPCZWBYRA-UHFFFAOYSA-N 0.000 description 1
- 229950000536 butamisole Drugs 0.000 description 1
- 229950005478 butamoxane Drugs 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 1
- VWYQKFLLGRBICZ-UHFFFAOYSA-N butanilicaine Chemical compound CCCCNCC(=O)NC1=C(C)C=CC=C1Cl VWYQKFLLGRBICZ-UHFFFAOYSA-N 0.000 description 1
- 229960001290 butanilicaine Drugs 0.000 description 1
- 229950003426 butanixin Drugs 0.000 description 1
- 229950000295 butanserin Drugs 0.000 description 1
- 229950001078 butantrone Drugs 0.000 description 1
- DVLBYTMYSMAKHP-UHFFFAOYSA-N butaperazine Chemical compound C12=CC(C(=O)CCC)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(C)CC1 DVLBYTMYSMAKHP-UHFFFAOYSA-N 0.000 description 1
- 229960000608 butaperazine Drugs 0.000 description 1
- 229950008654 butaprost Drugs 0.000 description 1
- WQDZHQZKJMNOAY-UHFFFAOYSA-N butaverine Chemical compound C=1C=CC=CC=1C(CC(=O)OCCCC)N1CCCCC1 WQDZHQZKJMNOAY-UHFFFAOYSA-N 0.000 description 1
- 229950010200 butaverine Drugs 0.000 description 1
- 229940070709 butedronate Drugs 0.000 description 1
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 1
- 229950005337 buterizine Drugs 0.000 description 1
- CKWHSYRZDLWQFV-UHFFFAOYSA-N butetamate Chemical compound CCN(CC)CCOC(=O)C(CC)C1=CC=CC=C1 CKWHSYRZDLWQFV-UHFFFAOYSA-N 0.000 description 1
- 229960003119 butetamate Drugs 0.000 description 1
- WDICTQVBXKADBP-UHFFFAOYSA-N butethamine Chemical compound CC(C)CNCCOC(=O)C1=CC=C(N)C=C1 WDICTQVBXKADBP-UHFFFAOYSA-N 0.000 description 1
- 229950009376 butethamine Drugs 0.000 description 1
- UULSXYSSHHRCQK-UHFFFAOYSA-N butibufen Chemical compound CCC(C(O)=O)C1=CC=C(CC(C)C)C=C1 UULSXYSSHHRCQK-UHFFFAOYSA-N 0.000 description 1
- 229960002973 butibufen Drugs 0.000 description 1
- GVNYSERWAKVROD-UHFFFAOYSA-N butidrine Chemical compound C1CCCC2=CC(C(O)CNC(C)CC)=CC=C21 GVNYSERWAKVROD-UHFFFAOYSA-N 0.000 description 1
- 229950003097 butidrine Drugs 0.000 description 1
- 229950001618 butikacin Drugs 0.000 description 1
- 229950009387 butilfenin Drugs 0.000 description 1
- YEFFVIUQXXNVGG-SJLPKXTDSA-N butinazocine Chemical compound C1C2=CC=C(O)C=C2[C@@]2(O)C(C)(C)[C@@H]1N(CCC#C)CC2 YEFFVIUQXXNVGG-SJLPKXTDSA-N 0.000 description 1
- 229950004480 butinazocine Drugs 0.000 description 1
- LWPXJPFOEPMIRG-UHFFFAOYSA-N butinoline Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(O)C#CCN1CCCC1 LWPXJPFOEPMIRG-UHFFFAOYSA-N 0.000 description 1
- 229950006174 butinoline Drugs 0.000 description 1
- 229950004527 butirosin Drugs 0.000 description 1
- 229950000699 butixirate Drugs 0.000 description 1
- HGBFRHCDYZJRAO-UHFFFAOYSA-N butizide Chemical compound ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)NC(CC(C)C)NC2=C1 HGBFRHCDYZJRAO-UHFFFAOYSA-N 0.000 description 1
- 229950008955 butizide Drugs 0.000 description 1
- ZKSIPEYIAHUPNM-ZEQRLZLVSA-N butobendine Chemical compound C([C@H](CC)N(C)CCN(C)[C@@H](CC)COC(=O)C=1C=C(OC)C(OC)=C(OC)C=1)OC(=O)C1=CC(OC)=C(OC)C(OC)=C1 ZKSIPEYIAHUPNM-ZEQRLZLVSA-N 0.000 description 1
- 229950001141 butobendine Drugs 0.000 description 1
- 229960005074 butoconazole Drugs 0.000 description 1
- SWLMUYACZKCSHZ-UHFFFAOYSA-N butoconazole Chemical compound C1=CC(Cl)=CC=C1CCC(SC=1C(=CC=CC=1Cl)Cl)CN1C=NC=C1 SWLMUYACZKCSHZ-UHFFFAOYSA-N 0.000 description 1
- 229950011621 butocrolol Drugs 0.000 description 1
- WTBQLHRVRKXSPX-UHFFFAOYSA-N butoctamide Chemical compound CCCCC(CC)CNC(=O)CC(C)O WTBQLHRVRKXSPX-UHFFFAOYSA-N 0.000 description 1
- 229950006519 butoctamide Drugs 0.000 description 1
- NMBNQRJDEPOXCP-UHFFFAOYSA-N butofilolol Chemical compound CCCC(=O)C1=CC(F)=CC=C1OCC(O)CNC(C)(C)C NMBNQRJDEPOXCP-UHFFFAOYSA-N 0.000 description 1
- 229950009191 butofilolol Drugs 0.000 description 1
- 229950010691 butonate Drugs 0.000 description 1
- 229950011520 butopamine Drugs 0.000 description 1
- 229950011442 butopiprine Drugs 0.000 description 1
- 229950007869 butoprozine Drugs 0.000 description 1
- 229950001472 butopyrammonium iodide Drugs 0.000 description 1
- IFKLAQQSCNILHL-QHAWAJNXSA-N butorphanol Chemical compound N1([C@@H]2CC3=CC=C(C=C3[C@@]3([C@]2(CCCC3)O)CC1)O)CC1CCC1 IFKLAQQSCNILHL-QHAWAJNXSA-N 0.000 description 1
- 229960001113 butorphanol Drugs 0.000 description 1
- 229950003621 butoxylate Drugs 0.000 description 1
- 229960004301 butriptyline Drugs 0.000 description 1
- ALELTFCQZDXAMQ-UHFFFAOYSA-N butriptyline Chemical compound C1CC2=CC=CC=C2C(CC(C)CN(C)C)C2=CC=CC=C21 ALELTFCQZDXAMQ-UHFFFAOYSA-N 0.000 description 1
- 229950008374 butropium bromide Drugs 0.000 description 1
- MQWDTXAOPTYTLC-UHFFFAOYSA-N butyl 1-(3-cyano-3,3-diphenylpropyl)-4-phenylpiperidine-4-carboxylate Chemical compound C1CC(C(=O)OCCCC)(C=2C=CC=CC=2)CCN1CCC(C#N)(C=1C=CC=CC=1)C1=CC=CC=C1 MQWDTXAOPTYTLC-UHFFFAOYSA-N 0.000 description 1
- YHLWPOLSPCBOPC-UHFFFAOYSA-O butyl(2-phosphopropan-2-yl)azanium Chemical compound CCCCNC(C)(C)[P+](O)=O YHLWPOLSPCBOPC-UHFFFAOYSA-O 0.000 description 1
- SZHRRALHWUGUQA-UHFFFAOYSA-M butyl-(1,5-dimethyl-3-oxo-2-phenylpyrazol-4-yl)-dimethylazanium;iodide Chemical compound [I-].O=C1C([N+](C)(C)CCCC)=C(C)N(C)N1C1=CC=CC=C1 SZHRRALHWUGUQA-UHFFFAOYSA-M 0.000 description 1
- 229940043253 butylated hydroxyanisole Drugs 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 1
- 229940067596 butylparaben Drugs 0.000 description 1
- PAVORIOUUILGMQ-UHFFFAOYSA-N butynamine Chemical compound CC(C)(C)N(C)C(C)(C)C#C PAVORIOUUILGMQ-UHFFFAOYSA-N 0.000 description 1
- 229950004968 butynamine Drugs 0.000 description 1
- 229960001336 buzepide metiodide Drugs 0.000 description 1
- HMFBCJNMIYZRKQ-UHFFFAOYSA-N buzepide metiodide Chemical compound [I-].C=1C=CC=CC=1C(C(N)=O)(C=1C=CC=CC=1)CC[N+]1(C)CCCCCC1 HMFBCJNMIYZRKQ-UHFFFAOYSA-N 0.000 description 1
- 229950003752 cabastine Drugs 0.000 description 1
- 229960004596 cabergoline Drugs 0.000 description 1
- 229960005211 cadralazine Drugs 0.000 description 1
- ZGNRRVAPHPANFI-UHFFFAOYSA-N cafaminol Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=C(N(CCO)C)N2C ZGNRRVAPHPANFI-UHFFFAOYSA-N 0.000 description 1
- 229950003668 cafaminol Drugs 0.000 description 1
- 229960004252 cafedrine Drugs 0.000 description 1
- UJSKUDDDPKGBJY-WFASDCNBSA-N cafedrine Chemical compound C1([C@@H](O)[C@@H](NCCN2C=3C(=O)N(C)C(=O)N(C)C=3N=C2)C)=CC=CC=C1 UJSKUDDDPKGBJY-WFASDCNBSA-N 0.000 description 1
- JWUBBDSIWDLEOM-DTOXIADCSA-N calcidiol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1\C[C@@H](O)CCC1=C JWUBBDSIWDLEOM-DTOXIADCSA-N 0.000 description 1
- 229960004361 calcifediol Drugs 0.000 description 1
- FAPWYRCQGJNNSJ-UBKPKTQASA-L calcium D-pantothenic acid Chemical compound [Ca+2].OCC(C)(C)[C@@H](O)C(=O)NCCC([O-])=O.OCC(C)(C)[C@@H](O)C(=O)NCCC([O-])=O FAPWYRCQGJNNSJ-UBKPKTQASA-L 0.000 description 1
- FNAQSUUGMSOBHW-UHFFFAOYSA-H calcium citrate Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O FNAQSUUGMSOBHW-UHFFFAOYSA-H 0.000 description 1
- 239000001354 calcium citrate Substances 0.000 description 1
- 229960004256 calcium citrate Drugs 0.000 description 1
- QGNBTYAQAPLTMX-UHFFFAOYSA-L calcium dobesilate Chemical compound [Ca+2].OC1=CC=C(O)C(S([O-])(=O)=O)=C1.OC1=CC=C(O)C(S([O-])(=O)=O)=C1 QGNBTYAQAPLTMX-UHFFFAOYSA-L 0.000 description 1
- 229960005438 calcium dobesilate Drugs 0.000 description 1
- YPCRNBPOUVJVMU-LCGAVOCYSA-L calcium glubionate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.[O-]C(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O YPCRNBPOUVJVMU-LCGAVOCYSA-L 0.000 description 1
- 229960002283 calcium glubionate Drugs 0.000 description 1
- 229940078512 calcium gluceptate Drugs 0.000 description 1
- 239000004227 calcium gluconate Substances 0.000 description 1
- 229960004494 calcium gluconate Drugs 0.000 description 1
- 235000013927 calcium gluconate Nutrition 0.000 description 1
- UHHRFSOMMCWGSO-UHFFFAOYSA-L calcium glycerophosphate Chemical compound [Ca+2].OCC(CO)OP([O-])([O-])=O UHHRFSOMMCWGSO-UHFFFAOYSA-L 0.000 description 1
- 229940095618 calcium glycerophosphate Drugs 0.000 description 1
- 235000019299 calcium glycerylphosphate Nutrition 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 229940064002 calcium hypophosphite Drugs 0.000 description 1
- 229910001382 calcium hypophosphite Inorganic materials 0.000 description 1
- MKJXYGKVIBWPFZ-UHFFFAOYSA-L calcium lactate Chemical compound [Ca+2].CC(O)C([O-])=O.CC(O)C([O-])=O MKJXYGKVIBWPFZ-UHFFFAOYSA-L 0.000 description 1
- 239000001527 calcium lactate Substances 0.000 description 1
- 235000011086 calcium lactate Nutrition 0.000 description 1
- 229960002401 calcium lactate Drugs 0.000 description 1
- 235000019307 calcium lactobionate Nutrition 0.000 description 1
- 229940050954 calcium lactobionate Drugs 0.000 description 1
- 229940078480 calcium levulinate Drugs 0.000 description 1
- 229960002079 calcium pantothenate Drugs 0.000 description 1
- 229940078495 calcium phosphate dibasic Drugs 0.000 description 1
- 229960001436 calcium saccharate Drugs 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 229940078456 calcium stearate Drugs 0.000 description 1
- RHEMCSSAABKPLI-SQCCMBKESA-L calcium;(2r,3r,4r,5r)-2,3,5,6-tetrahydroxy-4-[(2s,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyhexanoate Chemical compound [Ca+2].[O-]C(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O.[O-]C(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O RHEMCSSAABKPLI-SQCCMBKESA-L 0.000 description 1
- FATUQANACHZLRT-XBQZYUPDSA-L calcium;(2r,3r,4s,5r,6r)-2,3,4,5,6,7-hexahydroxyheptanoate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C([O-])=O FATUQANACHZLRT-XBQZYUPDSA-L 0.000 description 1
- UGZVNIRNPPEDHM-SBBOJQDXSA-L calcium;(2s,3s,4s,5r)-2,3,4,5-tetrahydroxyhexanedioate Chemical compound [Ca+2].[O-]C(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O UGZVNIRNPPEDHM-SBBOJQDXSA-L 0.000 description 1
- NEEHYRZPVYRGPP-UHFFFAOYSA-L calcium;2,3,4,5,6-pentahydroxyhexanoate Chemical compound [Ca+2].OCC(O)C(O)C(O)C(O)C([O-])=O.OCC(O)C(O)C(O)C(O)C([O-])=O NEEHYRZPVYRGPP-UHFFFAOYSA-L 0.000 description 1
- GCXSYHGQGJCRRW-UHFFFAOYSA-L calcium;2-hydroxy-2-phenylacetate Chemical compound [Ca+2].[O-]C(=O)C(O)C1=CC=CC=C1.[O-]C(=O)C(O)C1=CC=CC=C1 GCXSYHGQGJCRRW-UHFFFAOYSA-L 0.000 description 1
- IVFYLRMMHVYGJH-PVPPCFLZSA-N calusterone Chemical compound C1C[C@]2(C)[C@](O)(C)CC[C@H]2[C@@H]2[C@@H](C)CC3=CC(=O)CC[C@]3(C)[C@H]21 IVFYLRMMHVYGJH-PVPPCFLZSA-N 0.000 description 1
- 229950009823 calusterone Drugs 0.000 description 1
- 229960000926 camazepam Drugs 0.000 description 1
- PXBVEXGRHZFEOF-UHFFFAOYSA-N camazepam Chemical compound C12=CC(Cl)=CC=C2N(C)C(=O)C(OC(=O)N(C)C)N=C1C1=CC=CC=C1 PXBVEXGRHZFEOF-UHFFFAOYSA-N 0.000 description 1
- 229960003475 cambendazole Drugs 0.000 description 1
- 229950003586 camiverine Drugs 0.000 description 1
- 229950008603 camphotamide Drugs 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- 229960005242 camylofin Drugs 0.000 description 1
- 229950003720 canbisol Drugs 0.000 description 1
- UEKGZFCGRQYMRM-UHFFFAOYSA-N canbisol Chemical compound C1C(O)CCC2C(C)(C)OC3=CC(C(C)(C)CCCCCC)=CC(O)=C3C21 UEKGZFCGRQYMRM-UHFFFAOYSA-N 0.000 description 1
- HTQMVQVXFRQIKW-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=NN1 HTQMVQVXFRQIKW-UHFFFAOYSA-N 0.000 description 1
- 229960004349 candesartan cilexetil Drugs 0.000 description 1
- ZTGXAWYVTLUPDT-UHFFFAOYSA-N cannabidiol Natural products OC1=CC(CCCCC)=CC(O)=C1C1C(C(C)=C)CC=C(C)C1 ZTGXAWYVTLUPDT-UHFFFAOYSA-N 0.000 description 1
- 229960003453 cannabinol Drugs 0.000 description 1
- PBKZPPIHUVSDNM-WNHSNXHDSA-N canrenoic acid Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)CCC(O)=O)[C@@H]4[C@@H]3C=CC2=C1 PBKZPPIHUVSDNM-WNHSNXHDSA-N 0.000 description 1
- 229960002823 canrenoic acid Drugs 0.000 description 1
- UJVLDDZCTMKXJK-WNHSNXHDSA-N canrenone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CCC(=O)C=C3C=C2)C)CC[C@@]11C)C[C@@]11CCC(=O)O1 UJVLDDZCTMKXJK-WNHSNXHDSA-N 0.000 description 1
- 229960005057 canrenone Drugs 0.000 description 1
- DHZBEENLJMYSHQ-XCVPVQRUSA-N cantharidin Chemical compound C([C@@H]1O2)C[C@@H]2[C@]2(C)[C@@]1(C)C(=O)OC2=O DHZBEENLJMYSHQ-XCVPVQRUSA-N 0.000 description 1
- DHZBEENLJMYSHQ-UHFFFAOYSA-N cantharidine Natural products O1C2CCC1C1(C)C2(C)C(=O)OC1=O DHZBEENLJMYSHQ-UHFFFAOYSA-N 0.000 description 1
- 229950007443 capobenic acid Drugs 0.000 description 1
- 229960004602 capreomycin Drugs 0.000 description 1
- 229960002968 capreomycin sulfate Drugs 0.000 description 1
- 229950010003 caproxamine Drugs 0.000 description 1
- ZIWQJHATUBOOFD-SDXDJHTJSA-N caproxamine Chemical compound CCCCC\C(=N\OCCN)C1=CC=C(C)C(N)=C1 ZIWQJHATUBOOFD-SDXDJHTJSA-N 0.000 description 1
- 229960002504 capsaicin Drugs 0.000 description 1
- YKPUWZUDDOIDPM-SOFGYWHQSA-N capsaicin Chemical compound COC1=CC(CNC(=O)CCCC\C=C\C(C)C)=CC=C1O YKPUWZUDDOIDPM-SOFGYWHQSA-N 0.000 description 1
- 229950006417 captamine Drugs 0.000 description 1
- IZLPZXSZLLELBJ-UHFFFAOYSA-N captodiame Chemical compound C1=CC(SCCCC)=CC=C1C(SCCN(C)C)C1=CC=CC=C1 IZLPZXSZLLELBJ-UHFFFAOYSA-N 0.000 description 1
- 229960002574 captodiame Drugs 0.000 description 1
- HLSLSXBFTXUKCY-UHFFFAOYSA-N capuride Chemical compound CCC(C)C(CC)C(=O)NC(N)=O HLSLSXBFTXUKCY-UHFFFAOYSA-N 0.000 description 1
- 229950003152 capuride Drugs 0.000 description 1
- 229950009338 caracemide Drugs 0.000 description 1
- OFAIGZWCDGNZGT-UHFFFAOYSA-N caramiphen Chemical compound C=1C=CC=CC=1C1(C(=O)OCCN(CC)CC)CCCC1 OFAIGZWCDGNZGT-UHFFFAOYSA-N 0.000 description 1
- 229960004160 caramiphen Drugs 0.000 description 1
- BQXQGZPYHWWCEB-UHFFFAOYSA-N carazolol Chemical compound N1C2=CC=CC=C2C2=C1C=CC=C2OCC(O)CNC(C)C BQXQGZPYHWWCEB-UHFFFAOYSA-N 0.000 description 1
- 229960004634 carazolol Drugs 0.000 description 1
- 229960004484 carbachol Drugs 0.000 description 1
- AIXAANGOTKPUOY-UHFFFAOYSA-N carbachol Chemical compound [Cl-].C[N+](C)(C)CCOC(N)=O AIXAANGOTKPUOY-UHFFFAOYSA-N 0.000 description 1
- 229960000427 carbadox Drugs 0.000 description 1
- 229950004977 carbaldrate Drugs 0.000 description 1
- FFGPTBGBLSHEPO-UHFFFAOYSA-N carbamazepine Chemical compound C1=CC2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 FFGPTBGBLSHEPO-UHFFFAOYSA-N 0.000 description 1
- 229960000623 carbamazepine Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 235000013877 carbamide Nutrition 0.000 description 1
- 229940078916 carbamide peroxide Drugs 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 229950000776 carbarsone Drugs 0.000 description 1
- 229960005286 carbaryl Drugs 0.000 description 1
- CVXBEEMKQHEXEN-UHFFFAOYSA-N carbaryl Chemical compound C1=CC=C2C(OC(=O)NC)=CC=CC2=C1 CVXBEEMKQHEXEN-UHFFFAOYSA-N 0.000 description 1
- VYMUGTALCSPLDM-UHFFFAOYSA-L carbasalate calcium Chemical compound [Ca+2].NC(N)=O.CC(=O)OC1=CC=CC=C1C([O-])=O.CC(=O)OC1=CC=CC=C1C([O-])=O VYMUGTALCSPLDM-UHFFFAOYSA-L 0.000 description 1
- 229950009114 carbazeran Drugs 0.000 description 1
- XSXCZNVKFKNLPR-SDQBBNPISA-N carbazochrome Chemical compound NC(=O)N/N=C/1C(=O)C=C2N(C)CC(O)C2=C\1 XSXCZNVKFKNLPR-SDQBBNPISA-N 0.000 description 1
- 229960002631 carbazochrome Drugs 0.000 description 1
- 229950008741 carbazocine Drugs 0.000 description 1
- 229960003669 carbenicillin Drugs 0.000 description 1
- FPPNZSSZRUTDAP-UWFZAAFLSA-N carbenicillin Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)C(C(O)=O)C1=CC=CC=C1 FPPNZSSZRUTDAP-UWFZAAFLSA-N 0.000 description 1
- 229960000530 carbenoxolone Drugs 0.000 description 1
- OBZHEBDUNPOCJG-SZTGPWMUSA-N carbenoxolone Chemical compound C([C@H]1C2=CC(=O)[C@@H]34)[C@](C)(C(O)=O)CC[C@@]1(C)CC[C@@]2(C)[C@]4(C)CC[C@H]1[C@@]3(C)CC[C@@H](OC(=O)CCC(O)=O)C1(C)C OBZHEBDUNPOCJG-SZTGPWMUSA-N 0.000 description 1
- 229950002856 carbenzide Drugs 0.000 description 1
- MSIUIUDKTKNUET-UHFFFAOYSA-N carbenzide Chemical compound CCOC(=O)NNC(C)C1=CC=CC=C1 MSIUIUDKTKNUET-UHFFFAOYSA-N 0.000 description 1
- 229960004205 carbidopa Drugs 0.000 description 1
- TZFNLOMSOLWIDK-JTQLQIEISA-N carbidopa (anhydrous) Chemical compound NN[C@@](C(O)=O)(C)CC1=CC=C(O)C(O)=C1 TZFNLOMSOLWIDK-JTQLQIEISA-N 0.000 description 1
- 229950003365 carbifene Drugs 0.000 description 1
- NQIZDFMZAXUZCZ-UHFFFAOYSA-N carbifene Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(OCC)C(=O)N(C)CCN(C)CCC1=CC=CC=C1 NQIZDFMZAXUZCZ-UHFFFAOYSA-N 0.000 description 1
- 229960001704 carbimazole Drugs 0.000 description 1
- CFOYWRHIYXMDOT-UHFFFAOYSA-N carbimazole Chemical compound CCOC(=O)N1C=CN(C)C1=S CFOYWRHIYXMDOT-UHFFFAOYSA-N 0.000 description 1
- 229960000428 carbinoxamine Drugs 0.000 description 1
- OJFSXZCBGQGRNV-UHFFFAOYSA-N carbinoxamine Chemical compound C=1C=CC=NC=1C(OCCN(C)C)C1=CC=C(Cl)C=C1 OJFSXZCBGQGRNV-UHFFFAOYSA-N 0.000 description 1
- 229960004399 carbocisteine Drugs 0.000 description 1
- 229950003854 carbocloral Drugs 0.000 description 1
- ITMSAWKLJVGBIT-UHFFFAOYSA-N carbocloral Chemical compound CCOC(=O)NC(O)C(Cl)(Cl)Cl ITMSAWKLJVGBIT-UHFFFAOYSA-N 0.000 description 1
- 229960005003 carbocromen Drugs 0.000 description 1
- KLOIYEQEVSIOOO-UHFFFAOYSA-N carbocromen Chemical compound CC1=C(CCN(CC)CC)C(=O)OC2=CC(OCC(=O)OCC)=CC=C21 KLOIYEQEVSIOOO-UHFFFAOYSA-N 0.000 description 1
- 229950005130 carbofenotion Drugs 0.000 description 1
- 125000000837 carbohydrate group Chemical group 0.000 description 1
- 229950005779 carbomycin Drugs 0.000 description 1
- 229960003395 carboprost Drugs 0.000 description 1
- DLJKPYFALUEJCK-MRVZPHNRSA-N carboprost Chemical compound CCCCC[C@](C)(O)\C=C\[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C\CCCC(O)=O DLJKPYFALUEJCK-MRVZPHNRSA-N 0.000 description 1
- 229960002115 carboquone Drugs 0.000 description 1
- 150000007942 carboxylates Chemical group 0.000 description 1
- 229960001658 carbromal Drugs 0.000 description 1
- OPNPQXLQERQBBV-UHFFFAOYSA-N carbromal Chemical compound CCC(Br)(CC)C(=O)NC(N)=O OPNPQXLQERQBBV-UHFFFAOYSA-N 0.000 description 1
- ZWGPHQZXAPWKOV-UHFFFAOYSA-N carbubarb Chemical compound NC(=O)OCCC1(CCCC)C(=O)NC(=O)NC1=O ZWGPHQZXAPWKOV-UHFFFAOYSA-N 0.000 description 1
- 229950010247 carbubarb Drugs 0.000 description 1
- 229950004298 carburazepam Drugs 0.000 description 1
- HFFJORVBQWPILU-UHFFFAOYSA-N carburazepam Chemical compound NC(=O)N1CC(=O)N(C)C2=CC=C(Cl)C=C2C1C1=CC=CC=C1 HFFJORVBQWPILU-UHFFFAOYSA-N 0.000 description 1
- 229960003362 carbutamide Drugs 0.000 description 1
- VDTNNGKXZGSZIP-UHFFFAOYSA-N carbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(N)C=C1 VDTNNGKXZGSZIP-UHFFFAOYSA-N 0.000 description 1
- 229960001386 carbuterol Drugs 0.000 description 1
- KEMXXQOFIRIICG-UHFFFAOYSA-N carbuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(NC(N)=O)=C1 KEMXXQOFIRIICG-UHFFFAOYSA-N 0.000 description 1
- 229950004135 carcainium chloride Drugs 0.000 description 1
- 238000013155 cardiography Methods 0.000 description 1
- 229950010123 carebastine Drugs 0.000 description 1
- XGHOVGYJHWQGCC-UHFFFAOYSA-N carebastine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(=O)CCCN1CCC(OC(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 XGHOVGYJHWQGCC-UHFFFAOYSA-N 0.000 description 1
- 229950009852 carfenazine Drugs 0.000 description 1
- YDSDEBIZUNNPOB-UHFFFAOYSA-N carfentanil Chemical compound C1CN(CCC=2C=CC=CC=2)CCC1(C(=O)OC)N(C(=O)CC)C1=CC=CC=C1 YDSDEBIZUNNPOB-UHFFFAOYSA-N 0.000 description 1
- 229950004689 carfentanil Drugs 0.000 description 1
- MTTYXGPMZKRTCI-UHFFFAOYSA-N carfimate Chemical compound NC(=O)OC(C#C)C1=CC=CC=C1 MTTYXGPMZKRTCI-UHFFFAOYSA-N 0.000 description 1
- 229950001927 carfimate Drugs 0.000 description 1
- 229960000717 carindacillin Drugs 0.000 description 1
- JIRBAUWICKGBFE-MNRDOXJOSA-N carindacillin Chemical group N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)C(C(=O)OC=1C=C2CCCC2=CC=1)C1=CC=CC=C1 JIRBAUWICKGBFE-MNRDOXJOSA-N 0.000 description 1
- OFZCIYFFPZCNJE-UHFFFAOYSA-N carisoprodol Chemical compound NC(=O)OCC(C)(CCC)COC(=O)NC(C)C OFZCIYFFPZCNJE-UHFFFAOYSA-N 0.000 description 1
- 229960004587 carisoprodol Drugs 0.000 description 1
- 229950004799 carmantadine Drugs 0.000 description 1
- 229950001882 carmetizide Drugs 0.000 description 1
- XREUEWVEMYWFFA-CSKJXFQVSA-N carminomycin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XREUEWVEMYWFFA-CSKJXFQVSA-N 0.000 description 1
- XREUEWVEMYWFFA-UHFFFAOYSA-N carminomycin I Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XREUEWVEMYWFFA-UHFFFAOYSA-N 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 229950010848 carnidazole Drugs 0.000 description 1
- 229960004203 carnitine Drugs 0.000 description 1
- 229950007651 carocainide Drugs 0.000 description 1
- MSPRUJDUTKRMLM-UHFFFAOYSA-N caroverine Chemical compound O=C1N(CCN(CC)CC)C2=CC=CC=C2N=C1CC1=CC=C(OC)C=C1 MSPRUJDUTKRMLM-UHFFFAOYSA-N 0.000 description 1
- 229960003355 caroverine Drugs 0.000 description 1
- KYCBWEZLKCTALM-UHFFFAOYSA-N caroxazone Chemical compound C1=CC=C2OC(=O)N(CC(=O)N)CC2=C1 KYCBWEZLKCTALM-UHFFFAOYSA-N 0.000 description 1
- 229950006044 caroxazone Drugs 0.000 description 1
- IXZCNCWEDOHVLK-UHFFFAOYSA-N carperidine Chemical compound C=1C=CC=CC=1C1(C(=O)OCC)CCN(CCC(N)=O)CC1 IXZCNCWEDOHVLK-UHFFFAOYSA-N 0.000 description 1
- 229950001687 carperidine Drugs 0.000 description 1
- TVPJGGZLZLUPOB-SPIKMXEPSA-N carphenazine maleate Chemical compound OC(=O)\C=C/C(O)=O.OC(=O)\C=C/C(O)=O.C12=CC(C(=O)CC)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(CCO)CC1 TVPJGGZLZLUPOB-SPIKMXEPSA-N 0.000 description 1
- 229950000869 carpindolol Drugs 0.000 description 1
- 229960003184 carprofen Drugs 0.000 description 1
- PUXBGTOOZJQSKH-UHFFFAOYSA-N carprofen Chemical compound C1=C(Cl)C=C2C3=CC=C(C(C(O)=O)C)C=C3NC2=C1 PUXBGTOOZJQSKH-UHFFFAOYSA-N 0.000 description 1
- 229950003631 carpronium chloride Drugs 0.000 description 1
- RZMKWKZIJJNSLQ-UHFFFAOYSA-M carpronium chloride Chemical compound [Cl-].COC(=O)CCC[N+](C)(C)C RZMKWKZIJJNSLQ-UHFFFAOYSA-M 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- OAYRYNVEFFWSHK-UHFFFAOYSA-N carsalam Chemical compound C1=CC=C2OC(=O)NC(=O)C2=C1 OAYRYNVEFFWSHK-UHFFFAOYSA-N 0.000 description 1
- 229950004289 carsalam Drugs 0.000 description 1
- 229950007168 cartazolate Drugs 0.000 description 1
- IQNQAOGGWGCROX-UHFFFAOYSA-N cartazolate Chemical compound CCCCNC1=C(C(=O)OCC)C=NC2=C1C=NN2CC IQNQAOGGWGCROX-UHFFFAOYSA-N 0.000 description 1
- 229960001222 carteolol Drugs 0.000 description 1
- LWAFSWPYPHEXKX-UHFFFAOYSA-N carteolol Chemical compound N1C(=O)CCC2=C1C=CC=C2OCC(O)CNC(C)(C)C LWAFSWPYPHEXKX-UHFFFAOYSA-N 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 229950001725 carubicin Drugs 0.000 description 1
- 229960000662 carumonam Drugs 0.000 description 1
- UIMOJFJSJSIGLV-JNHMLNOCSA-N carumonam Chemical compound O=C1N(S(O)(=O)=O)[C@H](COC(=O)N)[C@@H]1NC(=O)C(=N/OCC(O)=O)\C1=CSC(N)=N1 UIMOJFJSJSIGLV-JNHMLNOCSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 229960000222 carzenide Drugs 0.000 description 1
- UCAGLBKTLXCODC-UHFFFAOYSA-N carzenide Chemical compound NS(=O)(=O)C1=CC=C(C(O)=O)C=C1 UCAGLBKTLXCODC-UHFFFAOYSA-N 0.000 description 1
- 229960003609 cathine Drugs 0.000 description 1
- DLNKOYKMWOXYQA-IONNQARKSA-N cathine Chemical compound C[C@H](N)[C@@H](O)C1=CC=CC=C1 DLNKOYKMWOXYQA-IONNQARKSA-N 0.000 description 1
- PUAQLLVFLMYYJJ-ZETCQYMHSA-N cathinone Chemical compound C[C@H](N)C(=O)C1=CC=CC=C1 PUAQLLVFLMYYJJ-ZETCQYMHSA-N 0.000 description 1
- 229950002698 cathinone Drugs 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 229960003972 cefacetrile Drugs 0.000 description 1
- RRYMAQUWDLIUPV-BXKDBHETSA-N cefacetrile Chemical compound S1CC(COC(=O)C)=C(C(O)=O)N2C(=O)[C@@H](NC(=O)CC#N)[C@@H]12 RRYMAQUWDLIUPV-BXKDBHETSA-N 0.000 description 1
- FUBBGQLTSCSAON-PBFPGSCMSA-N cefaloglycin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)COC(=O)C)C(O)=O)=CC=CC=C1 FUBBGQLTSCSAON-PBFPGSCMSA-N 0.000 description 1
- 229950004030 cefaloglycin Drugs 0.000 description 1
- GOFCPYKUMJBHBH-RHSMWYFYSA-N cefaloram Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC(=O)C)C(O)=O)C(=O)CC1=CC=CC=C1 GOFCPYKUMJBHBH-RHSMWYFYSA-N 0.000 description 1
- 229950001373 cefaloram Drugs 0.000 description 1
- 229960003866 cefaloridine Drugs 0.000 description 1
- CZTQZXZIADLWOZ-CRAIPNDOSA-N cefaloridine Chemical compound O=C([C@@H](NC(=O)CC=1SC=CC=1)[C@H]1SC2)N1C(C(=O)[O-])=C2C[N+]1=CC=CC=C1 CZTQZXZIADLWOZ-CRAIPNDOSA-N 0.000 description 1
- 229960000603 cefalotin Drugs 0.000 description 1
- XIURVHNZVLADCM-IUODEOHRSA-N cefalotin Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC(=O)C)C(O)=O)C(=O)CC1=CC=CS1 XIURVHNZVLADCM-IUODEOHRSA-N 0.000 description 1
- 229950000042 cefaparole Drugs 0.000 description 1
- 229960004350 cefapirin Drugs 0.000 description 1
- 229960002420 cefatrizine Drugs 0.000 description 1
- ACXMTAJLYQCRGF-PBFPGSCMSA-N cefatrizine Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)[C@H](N)C=2C=CC(O)=CC=2)CC=1CSC1=CN=N[N]1 ACXMTAJLYQCRGF-PBFPGSCMSA-N 0.000 description 1
- 229950004359 cefazaflur Drugs 0.000 description 1
- HGXLJRWXCXSEJO-GMSGAONNSA-N cefazaflur Chemical compound CN1N=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)CSC(F)(F)F)[C@H]2SC1 HGXLJRWXCXSEJO-GMSGAONNSA-N 0.000 description 1
- 229960005312 cefazedone Drugs 0.000 description 1
- VTLCNEGVSVJLDN-MLGOLLRUSA-N cefazedone Chemical compound S1C(C)=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)CN3C=C(Cl)C(=O)C(Cl)=C3)[C@H]2SC1 VTLCNEGVSVJLDN-MLGOLLRUSA-N 0.000 description 1
- 229960001139 cefazolin Drugs 0.000 description 1
- MLYYVTUWGNIJIB-BXKDBHETSA-N cefazolin Chemical compound S1C(C)=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)CN3N=NN=C3)[C@H]2SC1 MLYYVTUWGNIJIB-BXKDBHETSA-N 0.000 description 1
- 229960001817 cefbuperazone Drugs 0.000 description 1
- SMSRCGPDNDCXFR-CYWZMYCQSA-N cefbuperazone Chemical compound O=C1C(=O)N(CC)CCN1C(=O)N[C@H]([C@H](C)O)C(=O)N[C@]1(OC)C(=O)N2C(C(O)=O)=C(CSC=3N(N=NN=3)C)CS[C@@H]21 SMSRCGPDNDCXFR-CYWZMYCQSA-N 0.000 description 1
- 229950002706 cefcanel Drugs 0.000 description 1
- 229950008533 cefcanel daloxate Drugs 0.000 description 1
- 229950007281 cefedrolor Drugs 0.000 description 1
- 229950009347 cefempidone Drugs 0.000 description 1
- 229960002100 cefepime Drugs 0.000 description 1
- HVFLCNVBZFFHBT-ZKDACBOMSA-N cefepime Chemical compound S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1C[N+]1(C)CCCC1 HVFLCNVBZFFHBT-ZKDACBOMSA-N 0.000 description 1
- 229960004041 cefetamet Drugs 0.000 description 1
- MQLRYUCJDNBWMV-GHXIOONMSA-N cefetamet Chemical compound N([C@@H]1C(N2C(=C(C)CS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 MQLRYUCJDNBWMV-GHXIOONMSA-N 0.000 description 1
- 229950003098 cefetrizole Drugs 0.000 description 1
- 229960002129 cefixime Drugs 0.000 description 1
- OKBVVJOGVLARMR-QSWIMTSFSA-N cefixime Chemical compound S1C(N)=NC(C(=N\OCC(O)=O)\C(=O)N[C@@H]2C(N3C(=C(C=C)CS[C@@H]32)C(O)=O)=O)=C1 OKBVVJOGVLARMR-QSWIMTSFSA-N 0.000 description 1
- 229960003791 cefmenoxime Drugs 0.000 description 1
- HJJDBAOLQAWBMH-YCRCPZNHSA-N cefmenoxime Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NN=NN1C HJJDBAOLQAWBMH-YCRCPZNHSA-N 0.000 description 1
- 229950008733 cefmepidium chloride Drugs 0.000 description 1
- 229960003585 cefmetazole Drugs 0.000 description 1
- SNBUBQHDYVFSQF-HIFRSBDPSA-N cefmetazole Chemical compound S([C@@H]1[C@@](C(N1C=1C(O)=O)=O)(NC(=O)CSCC#N)OC)CC=1CSC1=NN=NN1C SNBUBQHDYVFSQF-HIFRSBDPSA-N 0.000 description 1
- 229960002025 cefminox Drugs 0.000 description 1
- JSDXOWVAHXDYCU-VXSYNFHWSA-N cefminox Chemical compound S([C@@H]1[C@@](C(N1C=1C(O)=O)=O)(NC(=O)CSC[C@@H](N)C(O)=O)OC)CC=1CSC1=NN=NN1C JSDXOWVAHXDYCU-VXSYNFHWSA-N 0.000 description 1
- 229960001958 cefodizime Drugs 0.000 description 1
- XDZKBRJLTGRPSS-BGZQYGJUSA-N cefodizime Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NC(C)=C(CC(O)=O)S1 XDZKBRJLTGRPSS-BGZQYGJUSA-N 0.000 description 1
- 229960004682 cefoperazone Drugs 0.000 description 1
- GCFBRXLSHGKWDP-XCGNWRKASA-N cefoperazone Chemical compound O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=CC(O)=CC=1)C(=O)N[C@@H]1C(=O)N2C(C(O)=O)=C(CSC=3N(N=NN=3)C)CS[C@@H]21 GCFBRXLSHGKWDP-XCGNWRKASA-N 0.000 description 1
- 229960004292 ceforanide Drugs 0.000 description 1
- SLAYUXIURFNXPG-CRAIPNDOSA-N ceforanide Chemical compound NCC1=CC=CC=C1CC(=O)N[C@@H]1C(=O)N2C(C(O)=O)=C(CSC=3N(N=NN=3)CC(O)=O)CS[C@@H]21 SLAYUXIURFNXPG-CRAIPNDOSA-N 0.000 description 1
- 229960004261 cefotaxime Drugs 0.000 description 1
- GPRBEKHLDVQUJE-VINNURBNSA-N cefotaxime Chemical compound N([C@@H]1C(N2C(=C(COC(C)=O)CS[C@@H]21)C(O)=O)=O)C(=O)/C(=N/OC)C1=CSC(N)=N1 GPRBEKHLDVQUJE-VINNURBNSA-N 0.000 description 1
- 229960005495 cefotetan Drugs 0.000 description 1
- SRZNHPXWXCNNDU-RHBCBLIFSA-N cefotetan Chemical compound N([C@]1(OC)C(N2C(=C(CSC=3N(N=NN=3)C)CS[C@@H]21)C(O)=O)=O)C(=O)C1SC(=C(C(N)=O)C(O)=O)S1 SRZNHPXWXCNNDU-RHBCBLIFSA-N 0.000 description 1
- 229960001242 cefotiam Drugs 0.000 description 1
- 229950002823 cefoxazole Drugs 0.000 description 1
- 229960002682 cefoxitin Drugs 0.000 description 1
- WZOZEZRFJCJXNZ-ZBFHGGJFSA-N cefoxitin Chemical compound N([C@]1(OC)C(N2C(=C(COC(N)=O)CS[C@@H]21)C(O)=O)=O)C(=O)CC1=CC=CS1 WZOZEZRFJCJXNZ-ZBFHGGJFSA-N 0.000 description 1
- LNZMRLHZGOBKAN-KAWPREARSA-N cefpimizole Chemical compound N1=CNC(C(=O)N[C@@H](C(=O)N[C@@H]2C(N3C(=C(C[N+]=4C=CC(CCS(O)(=O)=O)=CC=4)CS[C@@H]32)C([O-])=O)=O)C=2C=CC=CC=2)=C1C(=O)O LNZMRLHZGOBKAN-KAWPREARSA-N 0.000 description 1
- 229950004036 cefpimizole Drugs 0.000 description 1
- 229960005446 cefpiramide Drugs 0.000 description 1
- PWAUCHMQEXVFJR-PMAPCBKXSA-N cefpiramide Chemical compound C1=NC(C)=CC(O)=C1C(=O)N[C@H](C=1C=CC(O)=CC=1)C(=O)N[C@@H]1C(=O)N2C(C(O)=O)=C(CSC=3N(N=NN=3)C)CS[C@@H]21 PWAUCHMQEXVFJR-PMAPCBKXSA-N 0.000 description 1
- 229960000466 cefpirome Drugs 0.000 description 1
- DKOQGJHPHLTOJR-WHRDSVKCSA-N cefpirome Chemical compound N([C@@H]1C(N2C(=C(C[N+]=3C=4CCCC=4C=CC=3)CS[C@@H]21)C([O-])=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 DKOQGJHPHLTOJR-WHRDSVKCSA-N 0.000 description 1
- 229960005090 cefpodoxime Drugs 0.000 description 1
- WYUSVOMTXWRGEK-HBWVYFAYSA-N cefpodoxime Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC)C(O)=O)C(=O)C(=N/OC)\C1=CSC(N)=N1 WYUSVOMTXWRGEK-HBWVYFAYSA-N 0.000 description 1
- 229960004797 cefpodoxime proxetil Drugs 0.000 description 1
- LTINZAODLRIQIX-FBXRGJNPSA-N cefpodoxime proxetil Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC)C(=O)OC(C)OC(=O)OC(C)C)C(=O)C(=N/OC)\C1=CSC(N)=N1 LTINZAODLRIQIX-FBXRGJNPSA-N 0.000 description 1
- 229950009592 cefquinome Drugs 0.000 description 1
- 229950003685 cefrotil Drugs 0.000 description 1
- 229960003844 cefroxadine Drugs 0.000 description 1
- RDMOROXKXONCAL-UEKVPHQBSA-N cefroxadine Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)OC)C(O)=O)=CCC=CC1 RDMOROXKXONCAL-UEKVPHQBSA-N 0.000 description 1
- 229960003202 cefsulodin Drugs 0.000 description 1
- SYLKGLMBLAAGSC-QLVMHMETSA-N cefsulodin Chemical compound C1=CC(C(=O)N)=CC=[N+]1CC1=C(C([O-])=O)N2C(=O)[C@@H](NC(=O)[C@@H](C=3C=CC=CC=3)S(O)(=O)=O)[C@H]2SC1 SYLKGLMBLAAGSC-QLVMHMETSA-N 0.000 description 1
- OFKRKCHCYWQZLY-XHBSWPGZSA-N cefsumide Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=CC(NS(C)(=O)=O)=C1 OFKRKCHCYWQZLY-XHBSWPGZSA-N 0.000 description 1
- 229950010594 cefsumide Drugs 0.000 description 1
- 229960000484 ceftazidime Drugs 0.000 description 1
- ORFOPKXBNMVMKC-DWVKKRMSSA-N ceftazidime Chemical compound S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC(C)(C)C(O)=O)C=2N=C(N)SC=2)CC=1C[N+]1=CC=CC=C1 ORFOPKXBNMVMKC-DWVKKRMSSA-N 0.000 description 1
- 229950000679 cefteram Drugs 0.000 description 1
- 229960004366 ceftezole Drugs 0.000 description 1
- DZMVCVMFETWNIU-LDYMZIIASA-N ceftezole Chemical compound O=C([C@@H](NC(=O)CN1N=NN=C1)[C@H]1SC2)N1C(C(=O)O)=C2CSC1=NN=CS1 DZMVCVMFETWNIU-LDYMZIIASA-N 0.000 description 1
- 229960005229 ceftiofur Drugs 0.000 description 1
- ZBHXIWJRIFEVQY-IHMPYVIRSA-N ceftiofur Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC(=O)C1=CC=CO1 ZBHXIWJRIFEVQY-IHMPYVIRSA-N 0.000 description 1
- WJXAHFZIHLTPFR-JLRJEBFFSA-N ceftiolene Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1\C=C\SC1=NNC(=O)C(=O)N1CC=O WJXAHFZIHLTPFR-JLRJEBFFSA-N 0.000 description 1
- 229950008880 ceftiolene Drugs 0.000 description 1
- 229950007152 ceftioxide Drugs 0.000 description 1
- 229960001991 ceftizoxime Drugs 0.000 description 1
- NNULBSISHYWZJU-LLKWHZGFSA-N ceftizoxime Chemical compound N([C@@H]1C(N2C(=CCS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 NNULBSISHYWZJU-LLKWHZGFSA-N 0.000 description 1
- 229960004755 ceftriaxone Drugs 0.000 description 1
- VAAUVRVFOQPIGI-SPQHTLEESA-N ceftriaxone Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NC(=O)C(=O)NN1C VAAUVRVFOQPIGI-SPQHTLEESA-N 0.000 description 1
- 229950010799 cefuracetime Drugs 0.000 description 1
- 229960001668 cefuroxime Drugs 0.000 description 1
- JFPVXVDWJQMJEE-IZRZKJBUSA-N cefuroxime Chemical compound N([C@@H]1C(N2C(=C(COC(N)=O)CS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CC=CO1 JFPVXVDWJQMJEE-IZRZKJBUSA-N 0.000 description 1
- 229960002320 celiprolol Drugs 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 229950008877 cetaben Drugs 0.000 description 1
- UWCBNAVPISMFJZ-UHFFFAOYSA-N cetamolol Chemical compound CNC(=O)COC1=CC=CC=C1OCC(O)CNC(C)(C)C UWCBNAVPISMFJZ-UHFFFAOYSA-N 0.000 description 1
- 229950003205 cetamolol Drugs 0.000 description 1
- 229950006721 cethexonium chloride Drugs 0.000 description 1
- 229960003549 cetiedil Drugs 0.000 description 1
- 229960001803 cetirizine Drugs 0.000 description 1
- 229950010009 cetocycline Drugs 0.000 description 1
- 229950006185 cetohexazine Drugs 0.000 description 1
- YBROOZNJUDHTGE-QINSGFPZSA-N cetotiamine Chemical compound CCOC(=O)OCC\C(SC(=O)OCC)=C(/C)N(C=O)CC1=CN=C(C)N=C1N YBROOZNJUDHTGE-QINSGFPZSA-N 0.000 description 1
- 229950007625 cetotiamine Drugs 0.000 description 1
- GOAZMLKQUGCOPO-UHFFFAOYSA-N cetoxime Chemical compound C=1C=CC=CC=1N(CC(/N)=N/O)CC1=CC=CC=C1 GOAZMLKQUGCOPO-UHFFFAOYSA-N 0.000 description 1
- 229950011200 cetoxime Drugs 0.000 description 1
- FHRSHSOEWXUORL-HDJSIYSDSA-N cetraxate Chemical compound C1C[C@@H](C[NH3+])CC[C@@H]1C(=O)OC1=CC=C(CCC([O-])=O)C=C1 FHRSHSOEWXUORL-HDJSIYSDSA-N 0.000 description 1
- 229950009533 cetraxate Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 229950001084 chaulmosulfone Drugs 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 230000009920 chelation Effects 0.000 description 1
- JXPCRJDMUSNASY-WEMUOSSPSA-N chembl150283 Chemical compound C1CN(/C=N/CCCCCCCC)CCC1C(C=1C=CC=CC=1)C1=CC=CC=C1 JXPCRJDMUSNASY-WEMUOSSPSA-N 0.000 description 1
- JQXXHWHPUNPDRT-BQVAUQFYSA-N chembl1523493 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2C=NN1CCN(C)CC1 JQXXHWHPUNPDRT-BQVAUQFYSA-N 0.000 description 1
- UKFDTMNJMKWWNK-UHFFFAOYSA-N chembl2104165 Chemical compound C12=CC(Cl)=CC=C2N\C(=N\CC2CC2)C[N+]([O-])=C1C1=CC=CC=C1 UKFDTMNJMKWWNK-UHFFFAOYSA-N 0.000 description 1
- GQAWDMWLORTZKF-PMOLBWCYSA-N chembl2104242 Chemical compound C12=CC=CC=C2C=CC2=CC=CC=C2C1O[C@H](C1)C[C@H]2CC[C@@H]1N2C GQAWDMWLORTZKF-PMOLBWCYSA-N 0.000 description 1
- IAXUQWSLRKIRFR-SAABIXHNSA-N chembl2104696 Chemical compound C1C[C@@H](CNC(=N)N)CC[C@@H]1C(=O)OC1=CC=CC=C1C(=O)OCC1=CC=CC=C1 IAXUQWSLRKIRFR-SAABIXHNSA-N 0.000 description 1
- PSWOBQSIXLVPDV-CXUHLZMHSA-N chembl2105120 Chemical compound C1=C(O)C(OC)=CC(\C=N\NC(=O)C=2C=CN=CC=2)=C1 PSWOBQSIXLVPDV-CXUHLZMHSA-N 0.000 description 1
- TWOQVRDPCSGVLB-VAWYXSNFSA-N chembl2105962 Chemical compound CC(=O)\N=C1\SC=CN1CC(O)C1=CC=CS1 TWOQVRDPCSGVLB-VAWYXSNFSA-N 0.000 description 1
- NAWCPJNPSJBJGQ-PMOLBWCYSA-N chembl2106091 Chemical compound C12=CC=CC=C2CCC2=CC=CC=C2C1C(=O)O[C@H](C1)C[C@H]2CC[C@@H]1N2C NAWCPJNPSJBJGQ-PMOLBWCYSA-N 0.000 description 1
- GEDPQKGMHLHFOZ-XYPYZODXSA-N chembl2106163 Chemical compound C1C[C@@H](O)CC[C@@H]1NCC1=CC(Br)=CC(Br)=C1O GEDPQKGMHLHFOZ-XYPYZODXSA-N 0.000 description 1
- ASOJLQIBBYOFDE-HCHBIZCOSA-N chembl2106443 Chemical compound C(/[C@H]1O[C@H]([C@H]([C@H]1O)O)[C@H](C)[C@H](OC)C(/C)=C/C=C/CNC(=O)[C@@H](CC)[C@]1(O)[C@@H]([C@H](O[C@H]2[C@@H]([C@H](OC)[C@H](O[C@H]3[C@@H]([C@H](O)[C@@H](OC)[C@H](C)O3)OC)[C@@H](C)O2)O)C(C)(C)[C@H](\C=C\C=C\C)O1)O)=C\C=C\C=C(/C)C(=O)C1=C(O)C=CN(C)C1=O ASOJLQIBBYOFDE-HCHBIZCOSA-N 0.000 description 1
- ZAGFSRDBSQUHPR-YYYJYMDPSA-M chembl2106488 Chemical compound [Br-].O([C@H]1C[C@H]2CC[C@@H](C1)[N+]2(C)CC(C)C)C(=O)C(C=1C=CC=CC=1)C1CCCC1 ZAGFSRDBSQUHPR-YYYJYMDPSA-M 0.000 description 1
- XHKFBADIZIDYBU-MLFVOLAISA-N chembl2106658 Chemical compound N(/[C@H]1[C@@H]2N(C1=O)[C@H](C(S2)(C)C)C(=O)OC(C)OC(=O)OCC)=C\N1CCCCCC1 XHKFBADIZIDYBU-MLFVOLAISA-N 0.000 description 1
- WRNQYBXJRPAGNS-DQRAZIAOSA-N chembl2110888 Chemical compound C1CN(CCCCCC)CCN1C(=N/CC(C)C)\C1=CC=CC=C1 WRNQYBXJRPAGNS-DQRAZIAOSA-N 0.000 description 1
- PBKVEOSEPXMKDN-LZHUFOCISA-N chembl2311030 Chemical class CS(O)(=O)=O.CS(O)(=O)=O.CS(O)(=O)=O.CS(O)(=O)=O.C1=CC([C@H]2C[C@H](CN(C)[C@@H]2C2)C(=O)N[C@]3(C(=O)N4[C@H](C(N5CCC[C@H]5[C@]4(O)O3)=O)C(C)C)C(C)C)=C3C2=CNC3=C1.C1=CC([C@H]2C[C@H](CN(C)[C@@H]2C2)C(=O)N[C@]3(C(=O)N4[C@H](C(N5CCC[C@H]5[C@]4(O)O3)=O)C(C)CC)C(C)C)=C3C2=CNC3=C1.C1=CC([C@H]2C[C@H](CN(C)[C@@H]2C2)C(=O)N[C@]3(C(=O)N4[C@H](C(N5CCC[C@H]5[C@]4(O)O3)=O)CC(C)C)C(C)C)=C3C2=CNC3=C1.C([C@H]1C(=O)N2CCC[C@H]2[C@]2(O)O[C@](C(N21)=O)(NC(=O)[C@H]1CN(C)[C@H]2[C@@H](C=3C=CC=C4NC=C(C=34)C2)C1)C(C)C)C1=CC=CC=C1 PBKVEOSEPXMKDN-LZHUFOCISA-N 0.000 description 1
- KLLIVCPQDTYMLC-HDJSIYSDSA-N chembl292009 Chemical compound C1C[C@@H](C(C)(C)C)CC[C@@H]1CC1=C(O)C(=O)C2=CC=CC=C2C1=O KLLIVCPQDTYMLC-HDJSIYSDSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000013626 chemical specie Substances 0.000 description 1
- 229960001745 chiniofon Drugs 0.000 description 1
- 229940045110 chitosan Drugs 0.000 description 1
- ONAOIDNSINNZOA-UHFFFAOYSA-N chloral betaine Chemical compound OC(O)C(Cl)(Cl)Cl.C[N+](C)(C)CC([O-])=O ONAOIDNSINNZOA-UHFFFAOYSA-N 0.000 description 1
- 229940118803 chloral betaine Drugs 0.000 description 1
- QVFWZNCVPCJQOP-UHFFFAOYSA-N chloralodol Chemical compound CC(O)(C)CC(C)OC(O)C(Cl)(Cl)Cl QVFWZNCVPCJQOP-UHFFFAOYSA-N 0.000 description 1
- 229960005083 chloralodol Drugs 0.000 description 1
- OJYGBLRPYBAHRT-IPQSZEQASA-N chloralose Chemical compound O1[C@H](C(Cl)(Cl)Cl)O[C@@H]2[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]21 OJYGBLRPYBAHRT-IPQSZEQASA-N 0.000 description 1
- 229950009941 chloralose Drugs 0.000 description 1
- PXKHGMGELZGJQE-ILBGXUMGSA-N chloramphenicol palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](NC(=O)C(Cl)Cl)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 PXKHGMGELZGJQE-ILBGXUMGSA-N 0.000 description 1
- 229960001805 chloramphenicol palmitate Drugs 0.000 description 1
- 229960004357 chloramphenicol succinate Drugs 0.000 description 1
- YRZQHIVOIFJEEE-UHFFFAOYSA-N chlorazanil Chemical compound NC1=NC=NC(NC=2C=CC(Cl)=CC=2)=N1 YRZQHIVOIFJEEE-UHFFFAOYSA-N 0.000 description 1
- 229950002325 chlorazanil Drugs 0.000 description 1
- VEVSKUJZSMGTMM-UHFFFAOYSA-N chlorbenzoxamine Chemical compound CC1=CC=CC=C1CN1CCN(CCOC(C=2C=CC=CC=2)C=2C(=CC=CC=2)Cl)CC1 VEVSKUJZSMGTMM-UHFFFAOYSA-N 0.000 description 1
- 229960003078 chlorbenzoxamine Drugs 0.000 description 1
- STLZCUYBVPNYED-UHFFFAOYSA-N chlorbetamide Chemical compound OCCN(C(=O)C(Cl)Cl)CC1=CC=C(Cl)C=C1Cl STLZCUYBVPNYED-UHFFFAOYSA-N 0.000 description 1
- 229950008246 chlorbetamide Drugs 0.000 description 1
- 229960004831 chlorcyclizine Drugs 0.000 description 1
- 229960004782 chlordiazepoxide Drugs 0.000 description 1
- ANTSCNMPPGJYLG-UHFFFAOYSA-N chlordiazepoxide Chemical compound O=N=1CC(NC)=NC2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 ANTSCNMPPGJYLG-UHFFFAOYSA-N 0.000 description 1
- 229950008749 chlordimorine Drugs 0.000 description 1
- 229960003260 chlorhexidine Drugs 0.000 description 1
- DXXUGBPKQDTBQW-UHFFFAOYSA-L chlorisondamine Chemical compound [Cl-].[Cl-].ClC1=C(Cl)C(Cl)=C(Cl)C2=C1C[N+](CC[N+](C)(C)C)(C)C2 DXXUGBPKQDTBQW-UHFFFAOYSA-L 0.000 description 1
- 229950002565 chlorisondamine Drugs 0.000 description 1
- 229960001616 chlormadinone acetate Drugs 0.000 description 1
- BJFGVYCULWBXKF-UHFFFAOYSA-M chlormerodrin Chemical compound Cl[Hg]CC(OC)CNC(N)=O BJFGVYCULWBXKF-UHFFFAOYSA-M 0.000 description 1
- 229950002901 chlormerodrin Drugs 0.000 description 1
- WEQAYVWKMWHEJO-UHFFFAOYSA-N chlormezanone Chemical compound O=S1(=O)CCC(=O)N(C)C1C1=CC=C(Cl)C=C1 WEQAYVWKMWHEJO-UHFFFAOYSA-N 0.000 description 1
- 229960002810 chlormezanone Drugs 0.000 description 1
- 229960000849 chlormidazole Drugs 0.000 description 1
- WNAQOLSMVPFGTE-UHFFFAOYSA-N chlormidazole Chemical compound CC1=NC2=CC=CC=C2N1CC1=CC=C(Cl)C=C1 WNAQOLSMVPFGTE-UHFFFAOYSA-N 0.000 description 1
- NEHMKBQYUWJMIP-NJFSPNSNSA-N chloro(114C)methane Chemical compound [14CH3]Cl NEHMKBQYUWJMIP-NJFSPNSNSA-N 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- AOGYCOYQMAVAFD-UHFFFAOYSA-N chlorocarbonic acid Chemical class OC(Cl)=O AOGYCOYQMAVAFD-UHFFFAOYSA-N 0.000 description 1
- 229960002242 chlorocresol Drugs 0.000 description 1
- 235000019406 chloropentafluoroethane Nutrition 0.000 description 1
- 229960001448 chloropyramine Drugs 0.000 description 1
- XAEXSWVTEJHRMH-UHFFFAOYSA-N chloropyrilene Chemical compound C=1C=CC=NC=1N(CCN(C)C)CC1=CC=C(Cl)S1 XAEXSWVTEJHRMH-UHFFFAOYSA-N 0.000 description 1
- 229950005434 chloropyrilene Drugs 0.000 description 1
- 229950008335 chloroserpidine Drugs 0.000 description 1
- CYDMQBQPVICBEU-UHFFFAOYSA-N chlorotetracycline Natural products C1=CC(Cl)=C2C(O)(C)C3CC4C(N(C)C)C(O)=C(C(N)=O)C(=O)C4(O)C(O)=C3C(=O)C2=C1O CYDMQBQPVICBEU-UHFFFAOYSA-N 0.000 description 1
- 229960002155 chlorothiazide Drugs 0.000 description 1
- AFYPFACVUDMOHA-UHFFFAOYSA-N chlorotrifluoromethane Chemical compound FC(F)(F)Cl AFYPFACVUDMOHA-UHFFFAOYSA-N 0.000 description 1
- 229960004736 chloroxine Drugs 0.000 description 1
- WDFKMLRRRCGAKS-UHFFFAOYSA-N chloroxine Chemical compound C1=CN=C2C(O)=C(Cl)C=C(Cl)C2=C1 WDFKMLRRRCGAKS-UHFFFAOYSA-N 0.000 description 1
- 229960005443 chloroxylenol Drugs 0.000 description 1
- 229960001480 chlorozotocin Drugs 0.000 description 1
- SOYKEARSMXGVTM-UHFFFAOYSA-N chlorphenamine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Cl)C=C1 SOYKEARSMXGVTM-UHFFFAOYSA-N 0.000 description 1
- 229960003291 chlorphenamine Drugs 0.000 description 1
- 229960003993 chlorphenesin Drugs 0.000 description 1
- 229960004878 chlorphenesin carbamate Drugs 0.000 description 1
- SKPLBLUECSEIFO-UHFFFAOYSA-N chlorphenesin carbamate Chemical compound NC(=O)OCC(O)COC1=CC=C(Cl)C=C1 SKPLBLUECSEIFO-UHFFFAOYSA-N 0.000 description 1
- 229950007585 chlorphenoctium amsonate Drugs 0.000 description 1
- 229960003686 chlorphenoxamine Drugs 0.000 description 1
- KKHPNPMTPORSQE-UHFFFAOYSA-N chlorphenoxamine Chemical compound C=1C=C(Cl)C=CC=1C(C)(OCCN(C)C)C1=CC=CC=C1 KKHPNPMTPORSQE-UHFFFAOYSA-N 0.000 description 1
- 229950007046 chlorphentermine Drugs 0.000 description 1
- DBOUGBAQLIXZLV-UHFFFAOYSA-N chlorproethazine Chemical compound C1=C(Cl)C=C2N(CCCN(CC)CC)C3=CC=CC=C3SC2=C1 DBOUGBAQLIXZLV-UHFFFAOYSA-N 0.000 description 1
- 229960003030 chlorproethazine Drugs 0.000 description 1
- ISZNZKHCRKXXAU-UHFFFAOYSA-N chlorproguanil Chemical compound CC(C)\N=C(/N)N=C(N)NC1=CC=C(Cl)C(Cl)=C1 ISZNZKHCRKXXAU-UHFFFAOYSA-N 0.000 description 1
- 229950000764 chlorproguanil Drugs 0.000 description 1
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 1
- 229960001076 chlorpromazine Drugs 0.000 description 1
- 229960001761 chlorpropamide Drugs 0.000 description 1
- 229960001552 chlorprothixene Drugs 0.000 description 1
- 229960002172 chlorquinaldol Drugs 0.000 description 1
- GPTXWRGISTZRIO-UHFFFAOYSA-N chlorquinaldol Chemical compound ClC1=CC(Cl)=C(O)C2=NC(C)=CC=C21 GPTXWRGISTZRIO-UHFFFAOYSA-N 0.000 description 1
- 229960001523 chlortalidone Drugs 0.000 description 1
- 229960004475 chlortetracycline Drugs 0.000 description 1
- CYDMQBQPVICBEU-XRNKAMNCSA-N chlortetracycline Chemical compound C1=CC(Cl)=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O CYDMQBQPVICBEU-XRNKAMNCSA-N 0.000 description 1
- 235000019365 chlortetracycline Nutrition 0.000 description 1
- YEKMWXFHPZBZLR-UHFFFAOYSA-N chlorthenoxazine Chemical compound C1=CC=C2OC(CCCl)NC(=O)C2=C1 YEKMWXFHPZBZLR-UHFFFAOYSA-N 0.000 description 1
- 229950007438 chlorthenoxazine Drugs 0.000 description 1
- 229940107137 cholecystokinin Drugs 0.000 description 1
- 235000019416 cholic acid Nutrition 0.000 description 1
- 229960002471 cholic acid Drugs 0.000 description 1
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 1
- SUHOQUVVVLNYQR-MRVPVSSYSA-N choline alfoscerate Chemical compound C[N+](C)(C)CCOP([O-])(=O)OC[C@H](O)CO SUHOQUVVVLNYQR-MRVPVSSYSA-N 0.000 description 1
- 229960003178 choline chloride Drugs 0.000 description 1
- SGMZJAMFUVOLNK-UHFFFAOYSA-M choline chloride Chemical compound [Cl-].C[N+](C)(C)CCO SGMZJAMFUVOLNK-UHFFFAOYSA-M 0.000 description 1
- 229950002478 chromocarb Drugs 0.000 description 1
- 229950001703 ciadox Drugs 0.000 description 1
- 229950003453 ciamexon Drugs 0.000 description 1
- 229950004938 cianergoline Drugs 0.000 description 1
- LQXYCDLHSKICDY-UHFFFAOYSA-N cianopramine Chemical compound C1CC2=CC=C(C#N)C=C2N(CCCN(C)C)C2=CC=CC=C21 LQXYCDLHSKICDY-UHFFFAOYSA-N 0.000 description 1
- 229950001408 cianopramine Drugs 0.000 description 1
- 229950007238 ciapilome Drugs 0.000 description 1
- 229960004757 cibenzoline Drugs 0.000 description 1
- ARUGKOZUKWAXDS-SEWALLKFSA-N cicaprost Chemical compound C1\C(=C/COCC(O)=O)C[C@@H]2[C@@H](C#C[C@@H](O)[C@@H](C)CC#CCC)[C@H](O)C[C@@H]21 ARUGKOZUKWAXDS-SEWALLKFSA-N 0.000 description 1
- 229950000634 cicaprost Drugs 0.000 description 1
- 229950004947 cicarperone Drugs 0.000 description 1
- 229950005136 ciclactate Drugs 0.000 description 1
- AJNAKEPPGKJNKU-UHFFFAOYSA-N ciclafrine Chemical compound OC1=CC=CC(C2OC3(CCCCCC3)NC2)=C1 AJNAKEPPGKJNKU-UHFFFAOYSA-N 0.000 description 1
- 229950005558 ciclafrine Drugs 0.000 description 1
- VKQDZNZTPGLGFD-UHFFFAOYSA-N ciclazindol Chemical compound C12=NCCCN2C2=CC=CC=C2C1(O)C1=CC=CC(Cl)=C1 VKQDZNZTPGLGFD-UHFFFAOYSA-N 0.000 description 1
- 229950009468 ciclazindol Drugs 0.000 description 1
- 229960001932 cicletanine Drugs 0.000 description 1
- 229960001020 ciclobendazole Drugs 0.000 description 1
- OXLKOMYHDYVIDM-UHFFFAOYSA-N ciclobendazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1CC1 OXLKOMYHDYVIDM-UHFFFAOYSA-N 0.000 description 1
- 229960003025 ciclonicate Drugs 0.000 description 1
- GQSGZTBDVNUIQS-DGCLKSJQSA-N ciclonicate Chemical compound C1C(C)(C)C[C@H](C)C[C@H]1OC(=O)C1=CC=CN=C1 GQSGZTBDVNUIQS-DGCLKSJQSA-N 0.000 description 1
- 229950003212 ciclonium bromide Drugs 0.000 description 1
- 229960003749 ciclopirox Drugs 0.000 description 1
- SCKYRAXSEDYPSA-UHFFFAOYSA-N ciclopirox Chemical compound ON1C(=O)C=C(C)C=C1C1CCCCC1 SCKYRAXSEDYPSA-UHFFFAOYSA-N 0.000 description 1
- 229950005574 ciclopramine Drugs 0.000 description 1
- AEOOLRRTECSMIN-UHFFFAOYSA-N ciclopramine Chemical compound C12=CC=CC=C2CCC2=CC=CC3=C2N1CCC3NC AEOOLRRTECSMIN-UHFFFAOYSA-N 0.000 description 1
- 229950002545 cicloprofen Drugs 0.000 description 1
- 229950003775 cicloprolol Drugs 0.000 description 1
- 229950008471 ciclosidomine Drugs 0.000 description 1
- AJPLPOWGYORUIF-UHFFFAOYSA-N ciclosidomine Chemical compound C1CCCCC1C(/[O-])=N/C(ON=1)=C[N+]=1N1CCOCC1 AJPLPOWGYORUIF-UHFFFAOYSA-N 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- ZOSHXIXUCKESEG-UHFFFAOYSA-N ciclotizolam Chemical compound ClC1=CC=CC=C1C(C=1C=C(Br)SC=1N12)=NCC1=NN=C2C1CCCCC1 ZOSHXIXUCKESEG-UHFFFAOYSA-N 0.000 description 1
- 229950003501 ciclotizolam Drugs 0.000 description 1
- 229950001519 ciclotropium bromide Drugs 0.000 description 1
- 229950004498 cicloxolone Drugs 0.000 description 1
- SOESEFMFRCCMQV-QWAPGEGQSA-N cicortonide Chemical compound C1C(C#N)=C2C=C(OCCCl)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)COC(=O)C)[C@@]1(C)C[C@@H]2O SOESEFMFRCCMQV-QWAPGEGQSA-N 0.000 description 1
- 229950008136 cicortonide Drugs 0.000 description 1
- 229950008442 cidoxepin Drugs 0.000 description 1
- 229950007191 cifostodine Drugs 0.000 description 1
- YZFWTZACSRHJQD-UHFFFAOYSA-N ciglitazone Chemical compound C=1C=C(CC2C(NC(=O)S2)=O)C=CC=1OCC1(C)CCCCC1 YZFWTZACSRHJQD-UHFFFAOYSA-N 0.000 description 1
- 229950009226 ciglitazone Drugs 0.000 description 1
- 229950007521 ciheptolane Drugs 0.000 description 1
- SGEKLKJQLHJVDK-LJQANCHMSA-N ciladopa Chemical compound C1=C(OC)C(OC)=CC=C1[C@H](O)CN1CCN(C=2C(C=CC=CC=2)=O)CC1 SGEKLKJQLHJVDK-LJQANCHMSA-N 0.000 description 1
- 229950006515 ciladopa Drugs 0.000 description 1
- DHSUYTOATWAVLW-WFVMDLQDSA-N cilastatin Chemical compound CC1(C)C[C@@H]1C(=O)N\C(=C/CCCCSC[C@H](N)C(O)=O)C(O)=O DHSUYTOATWAVLW-WFVMDLQDSA-N 0.000 description 1
- 229960005025 cilazapril Drugs 0.000 description 1
- HHHKFGXWKKUNCY-FHWLQOOXSA-N cilazapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N2[C@@H](CCCN2CCC1)C(O)=O)=O)CC1=CC=CC=C1 HHHKFGXWKKUNCY-FHWLQOOXSA-N 0.000 description 1
- 229950010233 cilazaprilat Drugs 0.000 description 1
- 229950007728 cilobamine Drugs 0.000 description 1
- MQILJMOEUMZBHK-FIMMUYGNSA-N cilobamine Chemical compound C1([C@]2(O)C3CCC(CC3)[C@H]2NC(C)C)=CC=C(Cl)C(Cl)=C1 MQILJMOEUMZBHK-FIMMUYGNSA-N 0.000 description 1
- ZKZKCEAHVFVZDJ-MTUMARHDSA-N cilofungin Chemical compound C1=CC(OCCCCCCCC)=CC=C1C(=O)N[C@@H]1C(=O)N[C@@H]([C@@H](C)O)C(=O)N2C[C@H](O)C[C@H]2C(=O)N[C@@H]([C@H](O)[C@@H](O)C=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)N2C[C@H](C)[C@H](O)[C@H]2C(=O)N[C@H](O)[C@H](O)C1 ZKZKCEAHVFVZDJ-MTUMARHDSA-N 0.000 description 1
- 229950007664 cilofungin Drugs 0.000 description 1
- 108010090182 cilofungin Proteins 0.000 description 1
- 229950002934 cilostamide Drugs 0.000 description 1
- 229960004588 cilostazol Drugs 0.000 description 1
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 description 1
- 229950002340 ciltoprazine Drugs 0.000 description 1
- 229950010971 cimaterol Drugs 0.000 description 1
- BDHMBGHSWFWYSP-UHFFFAOYSA-N cimemoxin Chemical compound NNCC1CCCCC1 BDHMBGHSWFWYSP-UHFFFAOYSA-N 0.000 description 1
- 229950005199 cimemoxin Drugs 0.000 description 1
- 229950001268 cimepanol Drugs 0.000 description 1
- 229960001380 cimetidine Drugs 0.000 description 1
- CCGSUNCLSOWKJO-UHFFFAOYSA-N cimetidine Chemical compound N#CNC(=N/C)\NCCSCC1=NC=N[C]1C CCGSUNCLSOWKJO-UHFFFAOYSA-N 0.000 description 1
- 229960003705 cimetropium bromide Drugs 0.000 description 1
- WDURTRGFUGAJHA-MMQBYREUSA-M cimetropium bromide Chemical compound [Br-].C[N+]1([C@@H]2CC(C[C@H]1[C@@H]1O[C@@H]12)OC(=O)[C@@H](CO)C=1C=CC=CC=1)CC1CC1 WDURTRGFUGAJHA-MMQBYREUSA-M 0.000 description 1
- 229950001660 cimoxatone Drugs 0.000 description 1
- 229960001747 cinchocaine Drugs 0.000 description 1
- PUFQVTATUTYEAL-UHFFFAOYSA-N cinchocaine Chemical compound C1=CC=CC2=NC(OCCCC)=CC(C(=O)NCCN(CC)CC)=C21 PUFQVTATUTYEAL-UHFFFAOYSA-N 0.000 description 1
- KMPWYEUPVWOPIM-UHFFFAOYSA-N cinchonidine Natural products C1=CC=C2C(C(C3N4CCC(C(C4)C=C)C3)O)=CC=NC2=C1 KMPWYEUPVWOPIM-UHFFFAOYSA-N 0.000 description 1
- KMPWYEUPVWOPIM-LSOMNZGLSA-N cinchonine Chemical compound C1=CC=C2C([C@@H]([C@H]3N4CC[C@H]([C@H](C4)C=C)C3)O)=CC=NC2=C1 KMPWYEUPVWOPIM-LSOMNZGLSA-N 0.000 description 1
- 229960002468 cinchophen Drugs 0.000 description 1
- 229950001887 cinecromen Drugs 0.000 description 1
- 229960005233 cineole Drugs 0.000 description 1
- 229950010504 cinepaxadil Drugs 0.000 description 1
- 229960000352 cinepazet Drugs 0.000 description 1
- XDUOTWNXVDBCDY-SREVYHEPSA-N cinepazet Chemical compound C1CN(CC(=O)OCC)CCN1C(=O)\C=C/C1=CC(OC)=C(OC)C(OC)=C1 XDUOTWNXVDBCDY-SREVYHEPSA-N 0.000 description 1
- 229950007394 cinepazic acid Drugs 0.000 description 1
- RCUDFXMNPQNBDU-VOTSOKGWSA-N cinepazide Chemical compound COC1=C(OC)C(OC)=CC(\C=C\C(=O)N2CCN(CC(=O)N3CCCC3)CC2)=C1 RCUDFXMNPQNBDU-VOTSOKGWSA-N 0.000 description 1
- 229960004201 cinepazide Drugs 0.000 description 1
- 229950005724 cinfenine Drugs 0.000 description 1
- 229950008056 cinfenoac Drugs 0.000 description 1
- 229950010783 cinflumide Drugs 0.000 description 1
- 229950000136 cingestol Drugs 0.000 description 1
- ZDLBNXXKDMLZMF-UHFFFAOYSA-N cinitapride Chemical compound CCOC1=CC(N)=C([N+]([O-])=O)C=C1C(=O)NC1CCN(CC2CC=CCC2)CC1 ZDLBNXXKDMLZMF-UHFFFAOYSA-N 0.000 description 1
- 229960003875 cinitapride Drugs 0.000 description 1
- NKPPORKKCMYYTO-DHZHZOJOSA-N cinmetacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)\C=C\C1=CC=CC=C1 NKPPORKKCMYYTO-DHZHZOJOSA-N 0.000 description 1
- 229950011171 cinmetacin Drugs 0.000 description 1
- 229950001090 cinnamaverine Drugs 0.000 description 1
- 229960001750 cinnamedrine Drugs 0.000 description 1
- DERZBLKQOCDDDZ-JLHYYAGUSA-N cinnarizine Chemical compound C1CN(C(C=2C=CC=CC=2)C=2C=CC=CC=2)CCN1C\C=C\C1=CC=CC=C1 DERZBLKQOCDDDZ-JLHYYAGUSA-N 0.000 description 1
- 229960000876 cinnarizine Drugs 0.000 description 1
- 229950010278 cinnarizine clofibrate Drugs 0.000 description 1
- 229950004230 cinnofuradione Drugs 0.000 description 1
- MTRUDCIZADOOBK-UHFFFAOYSA-N cinnofuron Chemical compound O=C1N(C2=CC=CC=C2C2=CC=CC=C22)N2C(=O)C1CC1CCCO1 MTRUDCIZADOOBK-UHFFFAOYSA-N 0.000 description 1
- CWFRPHYKKQVWNI-HADAFWAXSA-N cinodine Chemical compound O([C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1NC(N)=N)NC(=O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]3N(C(=O)N[C@H]3[C@H](O)CO2)C(N)=O)CO1)NC(N)=O)C)C1=CC=C(\C=C\C(=O)NCCCNCCCCN)C=C1.O([C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1NC(N)=N)NC(=O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@H]3NC(=O)O[C@@H]3CO2)NC(N)=O)CO1)NC(N)=O)C)C1=CC=C(\C=C\C(=O)NCCCNCCCCN)C=C1 CWFRPHYKKQVWNI-HADAFWAXSA-N 0.000 description 1
- 229960002753 cinolazepam Drugs 0.000 description 1
- XAXMYHMKTCNRRZ-UHFFFAOYSA-N cinolazepam Chemical compound C12=CC(Cl)=CC=C2N(CCC#N)C(=O)C(O)N=C1C1=CC=CC=C1F XAXMYHMKTCNRRZ-UHFFFAOYSA-N 0.000 description 1
- 229950002192 cinoquidox Drugs 0.000 description 1
- CMDKPGRTAQVGFQ-RMKNXTFCSA-N cinoxate Chemical compound CCOCCOC(=O)\C=C\C1=CC=C(OC)C=C1 CMDKPGRTAQVGFQ-RMKNXTFCSA-N 0.000 description 1
- 229960001063 cinoxate Drugs 0.000 description 1
- 229950007299 cinoxolone Drugs 0.000 description 1
- 229950000031 cinperene Drugs 0.000 description 1
- 229950009472 cinprazole Drugs 0.000 description 1
- 229950010672 cinpropazide Drugs 0.000 description 1
- LDCXGZCEMNMWIL-VOTSOKGWSA-N cinromide Chemical compound CCNC(=O)\C=C\C1=CC=CC(Br)=C1 LDCXGZCEMNMWIL-VOTSOKGWSA-N 0.000 description 1
- 229950008460 cinromide Drugs 0.000 description 1
- 229950005716 ciprafamide Drugs 0.000 description 1
- KFIQKMINEHFZSM-HKUYNNGSSA-N ciprefadol Chemical compound OC1=CC=CC([C@]23[C@@H](CCCC2)CN(CC2CC2)CC3)=C1 KFIQKMINEHFZSM-HKUYNNGSSA-N 0.000 description 1
- 229950009302 ciprefadol Drugs 0.000 description 1
- 229950002649 ciprocinonide Drugs 0.000 description 1
- 229960002174 ciprofibrate Drugs 0.000 description 1
- KPSRODZRAIWAKH-UHFFFAOYSA-N ciprofibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1C1C(Cl)(Cl)C1 KPSRODZRAIWAKH-UHFFFAOYSA-N 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 229950000325 cipropride Drugs 0.000 description 1
- 229950002234 ciproquazone Drugs 0.000 description 1
- 229950009522 ciprostene Drugs 0.000 description 1
- UVTLONZTPXCUPU-ZNMIVQPWSA-N ciramadol Chemical compound C([C@@H]1[C@@H](N(C)C)C=2C=C(O)C=CC=2)CCC[C@H]1O UVTLONZTPXCUPU-ZNMIVQPWSA-N 0.000 description 1
- 229950007653 ciramadol Drugs 0.000 description 1
- 229950008137 cirazoline Drugs 0.000 description 1
- NJMYODHXAKYRHW-DVZOWYKESA-N cis-flupenthixol Chemical compound C1CN(CCO)CCN1CC\C=C\1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C2/1 NJMYODHXAKYRHW-DVZOWYKESA-N 0.000 description 1
- 229960005132 cisapride Drugs 0.000 description 1
- DCSUBABJRXZOMT-IRLDBZIGSA-N cisapride Chemical compound C([C@@H]([C@@H](CC1)NC(=O)C=2C(=CC(N)=C(Cl)C=2)OC)OC)N1CCCOC1=CC=C(F)C=C1 DCSUBABJRXZOMT-IRLDBZIGSA-N 0.000 description 1
- DCSUBABJRXZOMT-UHFFFAOYSA-N cisapride Natural products C1CC(NC(=O)C=2C(=CC(N)=C(Cl)C=2)OC)C(OC)CN1CCCOC1=CC=C(F)C=C1 DCSUBABJRXZOMT-UHFFFAOYSA-N 0.000 description 1
- 229950002162 cisconazole Drugs 0.000 description 1
- 229950010657 cismadinone Drugs 0.000 description 1
- 229950006146 cistinexine Drugs 0.000 description 1
- 229960001653 citalopram Drugs 0.000 description 1
- AGBTZJDOBMDLPR-UHFFFAOYSA-N citatepine Chemical compound C12=CC(C#N)=CC=C2SC2=CC=CC=C2C2=C1CCN(C)CC2 AGBTZJDOBMDLPR-UHFFFAOYSA-N 0.000 description 1
- 229950000902 citatepine Drugs 0.000 description 1
- 229950000567 citenamide Drugs 0.000 description 1
- 229950004773 citenazone Drugs 0.000 description 1
- 229960001284 citicoline Drugs 0.000 description 1
- 229950002110 clamidoxic acid Drugs 0.000 description 1
- 229950005158 clanfenur Drugs 0.000 description 1
- 229950003756 clanobutin Drugs 0.000 description 1
- VUPBWNXFSDRWJE-UHFFFAOYSA-N clanobutin Chemical compound C1=CC(OC)=CC=C1N(CCCC(O)=O)C(=O)C1=CC=C(Cl)C=C1 VUPBWNXFSDRWJE-UHFFFAOYSA-N 0.000 description 1
- 229950003839 clantifen Drugs 0.000 description 1
- 229960002626 clarithromycin Drugs 0.000 description 1
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 1
- 229960003324 clavulanic acid Drugs 0.000 description 1
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 description 1
- YAQKGZXXQNKEET-UHFFFAOYSA-N clazolam Chemical compound C1C(=O)N(C)C2=CC=C(Cl)C=C2C2C3=CC=CC=C3CCN21 YAQKGZXXQNKEET-UHFFFAOYSA-N 0.000 description 1
- 229950010761 clazolam Drugs 0.000 description 1
- 229950006911 clazolimine Drugs 0.000 description 1
- QUUTUGLQZLNABV-UHFFFAOYSA-N clazuril Chemical compound C1=CC(Cl)=CC=C1C(C#N)C1=CC=C(N2C(NC(=O)C=N2)=O)C=C1Cl QUUTUGLQZLNABV-UHFFFAOYSA-N 0.000 description 1
- 229950003606 clazuril Drugs 0.000 description 1
- 229960001791 clebopride Drugs 0.000 description 1
- 229960003801 clefamide Drugs 0.000 description 1
- ODCUSWJXZDHLKV-UHFFFAOYSA-N clefamide Chemical compound C1=CC(CN(CCO)C(=O)C(Cl)Cl)=CC=C1OC1=CC=C([N+]([O-])=O)C=C1 ODCUSWJXZDHLKV-UHFFFAOYSA-N 0.000 description 1
- 229960002881 clemastine Drugs 0.000 description 1
- YNNUSGIPVFPVBX-NHCUHLMSSA-N clemastine Chemical compound CN1CCC[C@@H]1CCO[C@@](C)(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 YNNUSGIPVFPVBX-NHCUHLMSSA-N 0.000 description 1
- 229950009328 clemeprol Drugs 0.000 description 1
- 229950002020 clemizole Drugs 0.000 description 1
- CJXAEXPPLWQRFR-UHFFFAOYSA-N clemizole Chemical compound C1=CC(Cl)=CC=C1CN1C2=CC=CC=C2N=C1CN1CCCC1 CJXAEXPPLWQRFR-UHFFFAOYSA-N 0.000 description 1
- 229960001117 clenbuterol Drugs 0.000 description 1
- STJMRWALKKWQGH-UHFFFAOYSA-N clenbuterol Chemical compound CC(C)(C)NCC(O)C1=CC(Cl)=C(N)C(Cl)=C1 STJMRWALKKWQGH-UHFFFAOYSA-N 0.000 description 1
- 229950004728 clenpirin Drugs 0.000 description 1
- 229950002559 cletoquine Drugs 0.000 description 1
- 229950005728 clibucaine Drugs 0.000 description 1
- 229950000849 clidafidine Drugs 0.000 description 1
- 229950010886 clidanac Drugs 0.000 description 1
- CHCISLOJADQUNQ-UHFFFAOYSA-N climazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1Cl CHCISLOJADQUNQ-UHFFFAOYSA-N 0.000 description 1
- 229950001490 climazolam Drugs 0.000 description 1
- 229960003344 climbazole Drugs 0.000 description 1
- 229950003544 climiqualine Drugs 0.000 description 1
- OYSKUZDIHNKWLV-PRUAPSLNSA-N clindamycin palmitate Chemical compound O1[C@H](SC)[C@H](OC(=O)CCCCCCCCCCCCCCC)[C@@H](O)[C@@H](O)[C@H]1[C@@H]([C@H](C)Cl)NC(=O)[C@H]1N(C)C[C@H](CCC)C1 OYSKUZDIHNKWLV-PRUAPSLNSA-N 0.000 description 1
- 229960004714 clindamycin palmitate Drugs 0.000 description 1
- 229960002291 clindamycin phosphate Drugs 0.000 description 1
- 239000012568 clinical material Substances 0.000 description 1
- 229950003072 clinofibrate Drugs 0.000 description 1
- OFSPDNIHQQKLPZ-HZJYTTRNSA-N clinolamide Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(=O)NC1CCCCC1 OFSPDNIHQQKLPZ-HZJYTTRNSA-N 0.000 description 1
- 229950003421 clinolamide Drugs 0.000 description 1
- 229960005228 clioquinol Drugs 0.000 description 1
- 229950010946 clioxanide Drugs 0.000 description 1
- 229950005384 cliprofen Drugs 0.000 description 1
- CXOXHMZGEKVPMT-UHFFFAOYSA-N clobazam Chemical compound O=C1CC(=O)N(C)C2=CC=C(Cl)C=C2N1C1=CC=CC=C1 CXOXHMZGEKVPMT-UHFFFAOYSA-N 0.000 description 1
- 229960001403 clobazam Drugs 0.000 description 1
- 229950003781 clobenoside Drugs 0.000 description 1
- UOYOWSGCMGEQHC-MSEXXDKFSA-N clobenoside Chemical compound CCCO[C@@H]1[C@@H](O)C(OCC)O[C@@H]1[C@H](OCC=1C=CC(Cl)=CC=1)COCC1=CC=C(Cl)C=C1 UOYOWSGCMGEQHC-MSEXXDKFSA-N 0.000 description 1
- IDWVKNARDDZONS-UHFFFAOYSA-N clobenzepam Chemical compound O=C1N(CCN(C)C)C2=CC=C(Cl)C=C2NC2=CC=CC=C21 IDWVKNARDDZONS-UHFFFAOYSA-N 0.000 description 1
- 229950006730 clobenzepam Drugs 0.000 description 1
- 229960002492 clobenzorex Drugs 0.000 description 1
- LRXXRIXDSAEIOR-ZDUSSCGKSA-N clobenzorex Chemical compound C([C@H](C)NCC=1C(=CC=CC=1)Cl)C1=CC=CC=C1 LRXXRIXDSAEIOR-ZDUSSCGKSA-N 0.000 description 1
- 229950005345 clobenztropine Drugs 0.000 description 1
- OCAXFDULERPAJM-LUQWZTMUSA-N clobenztropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 OCAXFDULERPAJM-LUQWZTMUSA-N 0.000 description 1
- CBGUOGMQLZIXBE-XGQKBEPLSA-N clobetasol propionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CCl)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CBGUOGMQLZIXBE-XGQKBEPLSA-N 0.000 description 1
- 229960004703 clobetasol propionate Drugs 0.000 description 1
- 229960005465 clobetasone butyrate Drugs 0.000 description 1
- 229960002735 clobutinol Drugs 0.000 description 1
- UGOFYAXVVVXMQT-UHFFFAOYSA-N clobuzarit Chemical compound C1=CC(COC(C)(C)C(O)=O)=CC=C1C1=CC=C(Cl)C=C1 UGOFYAXVVVXMQT-UHFFFAOYSA-N 0.000 description 1
- 229950007550 clobuzarit Drugs 0.000 description 1
- 229950002206 clocanfamide Drugs 0.000 description 1
- QAZKXHSIKKNOHH-UHFFFAOYSA-N clocapramine Chemical compound C1CN(CCCN2C3=CC(Cl)=CC=C3CCC3=CC=CC=C32)CCC1(C(=O)N)N1CCCCC1 QAZKXHSIKKNOHH-UHFFFAOYSA-N 0.000 description 1
- 229950001534 clocapramine Drugs 0.000 description 1
- 229950002660 clociguanil Drugs 0.000 description 1
- ZSQANMZWGKYDER-JXMROGBWSA-N clocinizine Chemical compound C1=CC(Cl)=CC=C1C(C=1C=CC=CC=1)N1CCN(C\C=C\C=2C=CC=CC=2)CC1 ZSQANMZWGKYDER-JXMROGBWSA-N 0.000 description 1
- 229960003826 clocinizine Drugs 0.000 description 1
- 229960001357 clocortolone pivalate Drugs 0.000 description 1
- SXYZQZLHAIHKKY-GSTUPEFVSA-N clocortolone pivalate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(Cl)[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)COC(=O)C(C)(C)C)[C@@]2(C)C[C@@H]1O SXYZQZLHAIHKKY-GSTUPEFVSA-N 0.000 description 1
- 229950002839 clocoumarol Drugs 0.000 description 1
- 229950003477 clodacaine Drugs 0.000 description 1
- 229950010571 clodanolene Drugs 0.000 description 1
- VOGJJBHRUDVEFM-UHFFFAOYSA-N clodantoin Chemical compound CCCCC(CC)C1NC(=O)N(SC(Cl)(Cl)Cl)C1=O VOGJJBHRUDVEFM-UHFFFAOYSA-N 0.000 description 1
- 229960004208 clodantoin Drugs 0.000 description 1
- 229950001159 clodazon Drugs 0.000 description 1
- 229950011320 clodoxopone Drugs 0.000 description 1
- 229960002286 clodronic acid Drugs 0.000 description 1
- ACSIXWWBWUQEHA-UHFFFAOYSA-N clodronic acid Chemical compound OP(O)(=O)C(Cl)(Cl)P(O)(O)=O ACSIXWWBWUQEHA-UHFFFAOYSA-N 0.000 description 1
- 229960004287 clofazimine Drugs 0.000 description 1
- WDQPAMHFFCXSNU-BGABXYSRSA-N clofazimine Chemical compound C12=CC=CC=C2N=C2C=C(NC=3C=CC(Cl)=CC=3)C(=N/C(C)C)/C=C2N1C1=CC=C(Cl)C=C1 WDQPAMHFFCXSNU-BGABXYSRSA-N 0.000 description 1
- 229960004472 clofedanol Drugs 0.000 description 1
- WRCHFMBCVFFYEQ-UHFFFAOYSA-N clofedanol Chemical compound C=1C=CC=C(Cl)C=1C(O)(CCN(C)C)C1=CC=CC=C1 WRCHFMBCVFFYEQ-UHFFFAOYSA-N 0.000 description 1
- 229950009130 clofenamic acid Drugs 0.000 description 1
- 229960002883 clofenamide Drugs 0.000 description 1
- FFCARBNHUSWRGK-UHFFFAOYSA-N clofenciclan Chemical compound C=1C=C(Cl)C=CC=1C1(OCCN(CC)CC)CCCCC1 FFCARBNHUSWRGK-UHFFFAOYSA-N 0.000 description 1
- 229950000229 clofenciclan Drugs 0.000 description 1
- 229950004832 clofenetamine Drugs 0.000 description 1
- 229960002643 clofenotane Drugs 0.000 description 1
- 229950007244 clofenoxyde Drugs 0.000 description 1
- 229950000512 clofenvinfos Drugs 0.000 description 1
- 229950000942 clofeverine Drugs 0.000 description 1
- UORGKWWJEQDTGX-UHFFFAOYSA-N clofexamide Chemical compound CCN(CC)CCNC(=O)COC1=CC=C(Cl)C=C1 UORGKWWJEQDTGX-UHFFFAOYSA-N 0.000 description 1
- 229960001417 clofexamide Drugs 0.000 description 1
- 229960003140 clofezone Drugs 0.000 description 1
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 1
- 229960001214 clofibrate Drugs 0.000 description 1
- TXCGAZHTZHNUAI-UHFFFAOYSA-N clofibric acid Chemical compound OC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 TXCGAZHTZHNUAI-UHFFFAOYSA-N 0.000 description 1
- 229950008441 clofibric acid Drugs 0.000 description 1
- CXQGFLBVUNUQIA-UHFFFAOYSA-N clofibride Chemical compound CN(C)C(=O)CCCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 CXQGFLBVUNUQIA-UHFFFAOYSA-N 0.000 description 1
- 229960005049 clofibride Drugs 0.000 description 1
- 229950000802 clofilium phosphate Drugs 0.000 description 1
- 229960003769 clofoctol Drugs 0.000 description 1
- TZWKUQDQKPYNLL-UHFFFAOYSA-N cloforex Chemical compound CCOC(=O)NC(C)(C)CC1=CC=C(Cl)C=C1 TZWKUQDQKPYNLL-UHFFFAOYSA-N 0.000 description 1
- 229950008294 cloforex Drugs 0.000 description 1
- 229950010124 clofurac Drugs 0.000 description 1
- 229950010752 cloguanamil Drugs 0.000 description 1
- JFRLWWDJCFYFSU-UHFFFAOYSA-N clomacran Chemical compound C1=C(Cl)C=C2C(CCCN(C)C)C3=CC=CC=C3NC2=C1 JFRLWWDJCFYFSU-UHFFFAOYSA-N 0.000 description 1
- 229950001885 clomacran Drugs 0.000 description 1
- DGMZLCLHHVYDIS-UHFFFAOYSA-N clometacin Chemical compound CC=1N(CC(O)=O)C2=CC(OC)=CC=C2C=1C(=O)C1=CC=C(Cl)C=C1 DGMZLCLHHVYDIS-UHFFFAOYSA-N 0.000 description 1
- 229950001647 clometacin Drugs 0.000 description 1
- 229960004414 clomethiazole Drugs 0.000 description 1
- JKXQBIZCQJLVOS-GSNLGQFWSA-N clometocillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C(OC)C1=CC=C(Cl)C(Cl)=C1 JKXQBIZCQJLVOS-GSNLGQFWSA-N 0.000 description 1
- 229960001351 clometocillin Drugs 0.000 description 1
- 229960003608 clomifene Drugs 0.000 description 1
- 229950008864 clomifenoxide Drugs 0.000 description 1
- HAHOPPGVHWVBRR-UHFFFAOYSA-N clominorex Chemical compound O1C(N)=NCC1C1=CC=C(Cl)C=C1 HAHOPPGVHWVBRR-UHFFFAOYSA-N 0.000 description 1
- 229950000352 clominorex Drugs 0.000 description 1
- 229960004606 clomipramine Drugs 0.000 description 1
- 229960004094 clomocycline Drugs 0.000 description 1
- BXVOHUQQUBSHLD-XCTBDMBQSA-N clomocycline Chemical compound C1=CC(Cl)=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(=O)C(=C(/O)NCO)/C(=O)[C@@]4(O)C(=O)C3=C(O)C2=C1O BXVOHUQQUBSHLD-XCTBDMBQSA-N 0.000 description 1
- 229950006376 clomoxir Drugs 0.000 description 1
- DGBIGWXXNGSACT-UHFFFAOYSA-N clonazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1Cl DGBIGWXXNGSACT-UHFFFAOYSA-N 0.000 description 1
- 229960003120 clonazepam Drugs 0.000 description 1
- 229950003003 clonazoline Drugs 0.000 description 1
- 229960002896 clonidine Drugs 0.000 description 1
- GPZLDQAEBHTMPG-UHFFFAOYSA-N clonitazene Chemical compound N=1C2=CC([N+]([O-])=O)=CC=C2N(CCN(CC)CC)C=1CC1=CC=C(Cl)C=C1 GPZLDQAEBHTMPG-UHFFFAOYSA-N 0.000 description 1
- 229950001604 clonitazene Drugs 0.000 description 1
- SUAJWTBTMNHVBZ-UHFFFAOYSA-N clonitrate Chemical compound [O-][N+](=O)OCC(CCl)O[N+]([O-])=O SUAJWTBTMNHVBZ-UHFFFAOYSA-N 0.000 description 1
- 229950004347 clonitrate Drugs 0.000 description 1
- 229950001923 clonixeril Drugs 0.000 description 1
- CLOMYZFHNHFSIQ-UHFFFAOYSA-N clonixin Chemical compound CC1=C(Cl)C=CC=C1NC1=NC=CC=C1C(O)=O CLOMYZFHNHFSIQ-UHFFFAOYSA-N 0.000 description 1
- 229960001209 clonixin Drugs 0.000 description 1
- 229960004070 clopamide Drugs 0.000 description 1
- 229960001184 clopenthixol Drugs 0.000 description 1
- 229960002544 cloperastine Drugs 0.000 description 1
- 229950000551 cloperidone Drugs 0.000 description 1
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 1
- 229960003009 clopidogrel Drugs 0.000 description 1
- 229960000731 clopidol Drugs 0.000 description 1
- JCZYXTVBWHAWLL-UHFFFAOYSA-N clopimozide Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)CCCN1CCC(N2C(NC3=CC(Cl)=CC=C32)=O)CC1 JCZYXTVBWHAWLL-UHFFFAOYSA-N 0.000 description 1
- 229950007971 clopimozide Drugs 0.000 description 1
- 229950004183 clopipazan Drugs 0.000 description 1
- SJCRQMUYEQHNTC-UHFFFAOYSA-N clopirac Chemical compound CC1=CC(CC(O)=O)=C(C)N1C1=CC=C(Cl)C=C1 SJCRQMUYEQHNTC-UHFFFAOYSA-N 0.000 description 1
- 229950009185 clopirac Drugs 0.000 description 1
- 229950000515 cloponone Drugs 0.000 description 1
- YTJIBEDMAQUYSZ-FDNPDPBUSA-N cloprednol Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3C=C(Cl)C2=C1 YTJIBEDMAQUYSZ-FDNPDPBUSA-N 0.000 description 1
- 229960002219 cloprednol Drugs 0.000 description 1
- 229960004409 cloprostenol Drugs 0.000 description 1
- 229950011409 cloprothiazole Drugs 0.000 description 1
- 229950006513 cloquinate Drugs 0.000 description 1
- 229950000492 cloquinozine Drugs 0.000 description 1
- 229950002214 cloracetadol Drugs 0.000 description 1
- 229960004893 cloranolol Drugs 0.000 description 1
- XYCMOTOFHFTUIU-UHFFFAOYSA-N cloranolol Chemical compound CC(C)(C)NCC(O)COC1=CC(Cl)=CC=C1Cl XYCMOTOFHFTUIU-UHFFFAOYSA-N 0.000 description 1
- 229960004362 clorazepate Drugs 0.000 description 1
- XDDJGVMJFWAHJX-UHFFFAOYSA-N clorazepic acid Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(C(=O)O)N=C1C1=CC=CC=C1 XDDJGVMJFWAHJX-UHFFFAOYSA-N 0.000 description 1
- 229960005315 clorexolone Drugs 0.000 description 1
- 229950002858 clorgiline Drugs 0.000 description 1
- BTFHLQRNAMSNLC-UHFFFAOYSA-N clorgyline Chemical compound C#CCN(C)CCCOC1=CC=C(Cl)C=C1Cl BTFHLQRNAMSNLC-UHFFFAOYSA-N 0.000 description 1
- 229960002571 cloricromen Drugs 0.000 description 1
- GYNNRVJJLAVVTQ-UHFFFAOYSA-N cloricromen Chemical compound CC1=C(CCN(CC)CC)C(=O)OC2=C(Cl)C(OCC(=O)OCC)=CC=C21 GYNNRVJJLAVVTQ-UHFFFAOYSA-N 0.000 description 1
- 229960001261 cloridarol Drugs 0.000 description 1
- KBFBRIPYVVGWRS-UHFFFAOYSA-N cloridarol Chemical compound C=1C2=CC=CC=C2OC=1C(O)C1=CC=C(Cl)C=C1 KBFBRIPYVVGWRS-UHFFFAOYSA-N 0.000 description 1
- 229950001794 clorindanic acid Drugs 0.000 description 1
- 229960001307 clorindione Drugs 0.000 description 1
- NJDUWAXIURWWLN-UHFFFAOYSA-N clorindione Chemical compound C1=CC(Cl)=CC=C1C1C(=O)C2=CC=CC=C2C1=O NJDUWAXIURWWLN-UHFFFAOYSA-N 0.000 description 1
- 229950005888 clormecaine Drugs 0.000 description 1
- 229950002239 cloroperone Drugs 0.000 description 1
- 229940110683 clorophene Drugs 0.000 description 1
- SONHVLIDLXLSOL-UHFFFAOYSA-N cloroqualone Chemical compound CCC1=NC2=CC=CC=C2C(=O)N1C1=C(Cl)C=CC=C1Cl SONHVLIDLXLSOL-UHFFFAOYSA-N 0.000 description 1
- 229950005517 cloroqualone Drugs 0.000 description 1
- 229950011192 clorotepine Drugs 0.000 description 1
- XRYLGRGAWQSVQW-UHFFFAOYSA-N clorotepine Chemical compound C1CN(C)CCN1C1C2=CC(Cl)=CC=C2SC2=CC=CC=C2C1 XRYLGRGAWQSVQW-UHFFFAOYSA-N 0.000 description 1
- 229950011462 clorprenaline Drugs 0.000 description 1
- 229960000275 clorsulon Drugs 0.000 description 1
- 229950000649 clortermine Drugs 0.000 description 1
- HXCXASJHZQXCKK-UHFFFAOYSA-N clortermine Chemical compound CC(C)(N)CC1=CC=CC=C1Cl HXCXASJHZQXCKK-UHFFFAOYSA-N 0.000 description 1
- 229950004178 closantel Drugs 0.000 description 1
- 229950010369 closiramine Drugs 0.000 description 1
- 229960001481 clostebol Drugs 0.000 description 1
- KCZCIYZKSLLNNH-FBPKJDBXSA-N clostebol Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1Cl KCZCIYZKSLLNNH-FBPKJDBXSA-N 0.000 description 1
- 229960003864 clotiapine Drugs 0.000 description 1
- 229960003622 clotiazepam Drugs 0.000 description 1
- 229950002278 clotioxone Drugs 0.000 description 1
- 229960004022 clotrimazole Drugs 0.000 description 1
- VNFPBHJOKIVQEB-UHFFFAOYSA-N clotrimazole Chemical compound ClC1=CC=CC=C1C(N1C=NC=C1)(C=1C=CC=CC=1)C1=CC=CC=C1 VNFPBHJOKIVQEB-UHFFFAOYSA-N 0.000 description 1
- 229950002663 clovoxamine Drugs 0.000 description 1
- XXPVSQRPGBUFKM-SAPNQHFASA-N clovoxamine Chemical compound COCCCC\C(=N/OCCN)C1=CC=C(Cl)C=C1 XXPVSQRPGBUFKM-SAPNQHFASA-N 0.000 description 1
- 229950009864 cloxacepride Drugs 0.000 description 1
- 229960003326 cloxacillin Drugs 0.000 description 1
- LQOLIRLGBULYKD-JKIFEVAISA-N cloxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1Cl LQOLIRLGBULYKD-JKIFEVAISA-N 0.000 description 1
- COCFKSXGORCFOW-VZHMHXRYSA-N cloxacillin benzathine Chemical compound C=1C=CC=CC=1C[NH2+]CC[NH2+]CC1=CC=CC=C1.N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C([O-])=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1Cl.N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C([O-])=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1Cl COCFKSXGORCFOW-VZHMHXRYSA-N 0.000 description 1
- 229960003932 cloxazolam Drugs 0.000 description 1
- ZIXNZOBDFKSQTC-UHFFFAOYSA-N cloxazolam Chemical compound C12=CC(Cl)=CC=C2NC(=O)CN2CCOC21C1=CC=CC=C1Cl ZIXNZOBDFKSQTC-UHFFFAOYSA-N 0.000 description 1
- 229950001232 cloxestradiol Drugs 0.000 description 1
- 229950011057 cloximate Drugs 0.000 description 1
- 229950003660 cloxiquine Drugs 0.000 description 1
- CTQMJYWDVABFRZ-UHFFFAOYSA-N cloxiquine Chemical compound C1=CN=C2C(O)=CC=C(Cl)C2=C1 CTQMJYWDVABFRZ-UHFFFAOYSA-N 0.000 description 1
- 229950008183 cloxotestosterone Drugs 0.000 description 1
- DNADMXUXHNLBKR-SIGPKOBDSA-N cloxotestosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)OC(O)C(Cl)(Cl)Cl)[C@@H]4[C@@H]3CCC2=C1 DNADMXUXHNLBKR-SIGPKOBDSA-N 0.000 description 1
- 229950008896 cloxypendyl Drugs 0.000 description 1
- 229960004170 clozapine Drugs 0.000 description 1
- QZUDBNBUXVUHMW-UHFFFAOYSA-N clozapine Chemical compound C1CN(C)CCN1C1=NC2=CC(Cl)=CC=C2NC2=CC=CC=C12 QZUDBNBUXVUHMW-UHFFFAOYSA-N 0.000 description 1
- 229960003920 cocaine Drugs 0.000 description 1
- 229950001485 cocarboxylase Drugs 0.000 description 1
- 229950002156 codoxime Drugs 0.000 description 1
- WKJYCUVUZIIMJA-OIBQYEROSA-N codoxime Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)C\C(=N/OCC(O)=O)[C@@H]1OC1=C2C3=CC=C1OC WKJYCUVUZIIMJA-OIBQYEROSA-N 0.000 description 1
- 229950000903 cofisatin Drugs 0.000 description 1
- 229950003743 cogazocine Drugs 0.000 description 1
- IUUBFDSJJHOTDI-UHFFFAOYSA-N cogazocine Chemical compound CC1(C)C2CC3=CC=C(O)C=C3C1(CC)CCN2CC1CCC1 IUUBFDSJJHOTDI-UHFFFAOYSA-N 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 229950010523 colestolone Drugs 0.000 description 1
- 229950003683 colfenamate Drugs 0.000 description 1
- 229940096422 collagen type i Drugs 0.000 description 1
- PHSMOUBHYUFTDM-UHFFFAOYSA-N colterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(O)=C1 PHSMOUBHYUFTDM-UHFFFAOYSA-N 0.000 description 1
- 229950004306 colterol Drugs 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- GPLGAQQQNWMVMM-MYAJQUOBSA-N conessine Chemical compound C1C=C2C[C@@H](N(C)C)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@H]3[C@H](C)N(C)C[C@@]31CC2 GPLGAQQQNWMVMM-MYAJQUOBSA-N 0.000 description 1
- 229950001827 conessine Drugs 0.000 description 1
- 238000002607 contrast-enhanced ultrasound Methods 0.000 description 1
- 229940108925 copper gluconate Drugs 0.000 description 1
- NHRTVBMNRNCBLQ-UHFFFAOYSA-L copper;5,7-disulfoquinolin-8-olate;n-ethylethanamine Chemical compound [Cu+2].CCNCC.CCNCC.CCNCC.CCNCC.C1=CC=NC2=C([O-])C(S(=O)(=O)O)=CC(S(O)(=O)=O)=C21.C1=CC=NC2=C([O-])C(S(=O)(=O)O)=CC(S(O)(=O)=O)=C21 NHRTVBMNRNCBLQ-UHFFFAOYSA-L 0.000 description 1
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 description 1
- ZKIDWWQMNXGYHV-BYJSBFAFSA-N cortisuzol Chemical compound O=C([C@]1(O)[C@@]2(C)C[C@H](O)[C@@H]3[C@@]4(C)CC5=C(N(N=C5)C=5C=CC=CC=5)C=C4C(C)=C[C@H]3[C@@H]2C[C@H]1C)COC(=O)C1=CC=CC(S(O)(=O)=O)=C1 ZKIDWWQMNXGYHV-BYJSBFAFSA-N 0.000 description 1
- 229950003515 cortisuzol Drugs 0.000 description 1
- 229960003840 cortivazol Drugs 0.000 description 1
- RKHQGWMMUURILY-UHRZLXHJSA-N cortivazol Chemical compound C([C@H]1[C@@H]2C[C@H]([C@]([C@@]2(C)C[C@H](O)[C@@H]1[C@@]1(C)C2)(O)C(=O)COC(C)=O)C)=C(C)C1=CC1=C2C=NN1C1=CC=CC=C1 RKHQGWMMUURILY-UHRZLXHJSA-N 0.000 description 1
- 229950002276 cortodoxone Drugs 0.000 description 1
- 229950006073 cotinine Drugs 0.000 description 1
- DRNINDKQWDOFMO-UHFFFAOYSA-N cotriptyline Chemical compound C1CC2=CC=CC=C2C(=CC(=O)CN(C)C)C2=CC=CC=C21 DRNINDKQWDOFMO-UHFFFAOYSA-N 0.000 description 1
- 229950010872 cotriptyline Drugs 0.000 description 1
- BXNANOICGRISHX-UHFFFAOYSA-N coumaphos Chemical compound CC1=C(Cl)C(=O)OC2=CC(OP(=S)(OCC)OCC)=CC=C21 BXNANOICGRISHX-UHFFFAOYSA-N 0.000 description 1
- 229950003525 coumazoline Drugs 0.000 description 1
- 229950001111 coumetarol Drugs 0.000 description 1
- BUCJFFQZPGTGPX-UHFFFAOYSA-N coumetarol Chemical compound C1=CC=C2OC(=O)C(C(C=3C(OC4=CC=CC=C4C=3O)=O)COC)=C(O)C2=C1 BUCJFFQZPGTGPX-UHFFFAOYSA-N 0.000 description 1
- 229960002956 creatinolfosfate Drugs 0.000 description 1
- FOIPWTMKYXWFGC-UHFFFAOYSA-N creatinolfosfate Chemical compound NC(=N)N(C)CCOP(O)(O)=O FOIPWTMKYXWFGC-UHFFFAOYSA-N 0.000 description 1
- SBRXTSOCZITGQG-UHFFFAOYSA-N crisnatol Chemical compound C1=CC=C2C(CNC(CO)(CO)C)=CC3=C(C=CC=C4)C4=CC=C3C2=C1 SBRXTSOCZITGQG-UHFFFAOYSA-N 0.000 description 1
- 229950007258 crisnatol Drugs 0.000 description 1
- 229960002042 croconazole Drugs 0.000 description 1
- 229950004210 cromakalim Drugs 0.000 description 1
- 229950002807 cromitrile Drugs 0.000 description 1
- 229960000265 cromoglicic acid Drugs 0.000 description 1
- 229950008982 cropropamide Drugs 0.000 description 1
- CYZWCBZIBJLKCV-RMKNXTFCSA-N cropropamide Chemical compound CN(C)C(=O)C(CC)N(CCC)C(=O)\C=C\C CYZWCBZIBJLKCV-RMKNXTFCSA-N 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 229960003338 crotamiton Drugs 0.000 description 1
- DNTGGZPQPQTDQF-XBXARRHUSA-N crotamiton Chemical compound C/C=C/C(=O)N(CC)C1=CC=CC=C1C DNTGGZPQPQTDQF-XBXARRHUSA-N 0.000 description 1
- 229950008678 crotetamide Drugs 0.000 description 1
- LSAMUAYPDHUBQD-RMKNXTFCSA-N crotetamide Chemical compound CN(C)C(=O)C(CC)N(CC)C(=O)\C=C\C LSAMUAYPDHUBQD-RMKNXTFCSA-N 0.000 description 1
- 229950000400 crotoniazide Drugs 0.000 description 1
- 229950002363 crufomate Drugs 0.000 description 1
- ZXJXZNDDNMQXFV-UHFFFAOYSA-M crystal violet Chemical compound [Cl-].C1=CC(N(C)C)=CC=C1[C+](C=1C=CC(=CC=1)N(C)C)C1=CC=C(N(C)C)C=C1 ZXJXZNDDNMQXFV-UHFFFAOYSA-M 0.000 description 1
- CFCXQNRBXWYPRI-UHFFFAOYSA-M cuprimyxin Chemical compound [Cu+2].C1=CC=C2[N+]([O-])=C3C(OC)=CC=CC3=[N+]([O-])C2=C1[O-] CFCXQNRBXWYPRI-UHFFFAOYSA-M 0.000 description 1
- 229950003458 cuprimyxin Drugs 0.000 description 1
- 229950009004 cuproxoline Drugs 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 229950000525 cyacetacide Drugs 0.000 description 1
- 229960004278 cyamemazine Drugs 0.000 description 1
- SLFGIOIONGJGRT-UHFFFAOYSA-N cyamemazine Chemical compound C1=C(C#N)C=C2N(CC(CN(C)C)C)C3=CC=CC=C3SC2=C1 SLFGIOIONGJGRT-UHFFFAOYSA-N 0.000 description 1
- NLCKLZIHJQEMCU-UHFFFAOYSA-N cyano prop-2-enoate Chemical class C=CC(=O)OC#N NLCKLZIHJQEMCU-UHFFFAOYSA-N 0.000 description 1
- 229960002104 cyanocobalamin Drugs 0.000 description 1
- 229960000729 cyclandelate Drugs 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- IRZVVDMCEZNNCW-UHFFFAOYSA-N cyclarbamate Chemical compound C1CCCC1(COC(=O)NC=1C=CC=CC=1)COC(=O)NC1=CC=CC=C1 IRZVVDMCEZNNCW-UHFFFAOYSA-N 0.000 description 1
- 229950006085 cyclarbamate Drugs 0.000 description 1
- 229950002213 cyclazocine Drugs 0.000 description 1
- DNRKTAYPGADPGW-UHFFFAOYSA-N cyclazodone Chemical compound O1C(C=2C=CC=CC=2)C(=O)N=C1NC1CC1 DNRKTAYPGADPGW-UHFFFAOYSA-N 0.000 description 1
- 229950007404 cyclazodone Drugs 0.000 description 1
- 229950004192 cyclexanone Drugs 0.000 description 1
- KUOYUNPLUMKQIF-UHFFFAOYSA-N cyclexanone Chemical compound O=C1CCCC1(C=1CCCC=1)CCN1CCOCC1 KUOYUNPLUMKQIF-UHFFFAOYSA-N 0.000 description 1
- 150000001923 cyclic compounds Chemical group 0.000 description 1
- 229950003459 cycliramine Drugs 0.000 description 1
- 229960003564 cyclizine Drugs 0.000 description 1
- UVKZSORBKUEBAZ-UHFFFAOYSA-N cyclizine Chemical compound C1CN(C)CCN1C(C=1C=CC=CC=1)C1=CC=CC=C1 UVKZSORBKUEBAZ-UHFFFAOYSA-N 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 229960004138 cyclobarbital Drugs 0.000 description 1
- WTYGAUXICFETTC-UHFFFAOYSA-N cyclobarbital Chemical compound C=1CCCCC=1C1(CC)C(=O)NC(=O)NC1=O WTYGAUXICFETTC-UHFFFAOYSA-N 0.000 description 1
- 229960003572 cyclobenzaprine Drugs 0.000 description 1
- JURKNVYFZMSNLP-UHFFFAOYSA-N cyclobenzaprine Chemical compound C1=CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 JURKNVYFZMSNLP-UHFFFAOYSA-N 0.000 description 1
- 229950003148 cyclobutoic acid Drugs 0.000 description 1
- 229960001752 cyclobutyrol Drugs 0.000 description 1
- NIVFTEMPSCMWDE-UHFFFAOYSA-N cyclobutyrol Chemical compound CCC(C(O)=O)C1(O)CCCCC1 NIVFTEMPSCMWDE-UHFFFAOYSA-N 0.000 description 1
- 229960002944 cyclofenil Drugs 0.000 description 1
- 229950004734 cycloguanil Drugs 0.000 description 1
- OCHNIEZVJROLLV-UHFFFAOYSA-N cyclohexyl 4-[4-(4-fluorophenyl)-4-oxobutyl]piperazine-1-carboxylate Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCN(C(=O)OC2CCCCC2)CC1 OCHNIEZVJROLLV-UHFFFAOYSA-N 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 229950005118 cyclomenol Drugs 0.000 description 1
- 229940086555 cyclomethicone Drugs 0.000 description 1
- YLRNESBGEGGQBK-UHFFFAOYSA-N cyclomethycaine Chemical compound CC1CCCCN1CCCOC(=O)C(C=C1)=CC=C1OC1CCCCC1 YLRNESBGEGGQBK-UHFFFAOYSA-N 0.000 description 1
- 229960004741 cyclomethycaine Drugs 0.000 description 1
- ACNAXEZQQXXWAE-UHFFFAOYSA-M cyclopenta-1,3-diene;1-cyclopenta-2,4-dien-1-ylidene-3,5,5-trimethylhexan-1-olate;iron(2+) Chemical compound [Fe+2].C=1C=C[CH-]C=1.CC(C)(C)CC(C)CC([O-])=C1C=CC=C1 ACNAXEZQQXXWAE-UHFFFAOYSA-M 0.000 description 1
- 229960003263 cyclopentamine Drugs 0.000 description 1
- HFXKQSZZZPGLKQ-UHFFFAOYSA-N cyclopentamine Chemical compound CNC(C)CC1CCCC1 HFXKQSZZZPGLKQ-UHFFFAOYSA-N 0.000 description 1
- 229960003206 cyclopenthiazide Drugs 0.000 description 1
- SKYSRIRYMSLOIN-UHFFFAOYSA-N cyclopentolate Chemical compound C1CCCC1(O)C(C(=O)OCCN(C)C)C1=CC=CC=C1 SKYSRIRYMSLOIN-UHFFFAOYSA-N 0.000 description 1
- 229960001815 cyclopentolate Drugs 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- UGIARLNNAPRCPF-HUPPADNKSA-N cyclopregnol Chemical compound C([C@H]1[C@@H]2CC[C@@H]([C@]2(CC[C@@H]1[C@@]1(C)CC2)C)C(=O)C)[C@@H](O)[C@]31[C@@H]2C3 UGIARLNNAPRCPF-HUPPADNKSA-N 0.000 description 1
- 229950002928 cyclopregnol Drugs 0.000 description 1
- 229950007502 cyclopyrronium bromide Drugs 0.000 description 1
- 229960003077 cycloserine Drugs 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 229960003176 cyclothiazide Drugs 0.000 description 1
- BOCUKUHCLICSIY-QJWLJZLASA-N cyclothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2C1[C@H](C=C2)C[C@H]2C1 BOCUKUHCLICSIY-QJWLJZLASA-N 0.000 description 1
- 229950006648 cyclovalone Drugs 0.000 description 1
- 229950000191 cycloxilic acid Drugs 0.000 description 1
- 229950002217 cycotiamine Drugs 0.000 description 1
- 229960000512 cycrimine Drugs 0.000 description 1
- SWRUZBWLEWHWRI-UHFFFAOYSA-N cycrimine Chemical compound C1CCCC1C(C=1C=CC=CC=1)(O)CCN1CCCCC1 SWRUZBWLEWHWRI-UHFFFAOYSA-N 0.000 description 1
- 229950002092 cyheptropine Drugs 0.000 description 1
- 229950009125 cynarine Drugs 0.000 description 1
- YDDUMTOHNYZQPO-BKUKFAEQSA-N cynarine Natural products O[C@H]1C[C@@](C[C@H](OC(=O)C=Cc2ccc(O)c(O)c2)[C@@H]1O)(OC(=O)C=Cc3ccc(O)c(O)c3)C(=O)O YDDUMTOHNYZQPO-BKUKFAEQSA-N 0.000 description 1
- VNGYTYNUZHDMPP-MNOVXSKESA-N cypenamine Chemical compound N[C@H]1CCC[C@@H]1C1=CC=CC=C1 VNGYTYNUZHDMPP-MNOVXSKESA-N 0.000 description 1
- 229950004202 cypenamine Drugs 0.000 description 1
- 229950010040 cyprazepam Drugs 0.000 description 1
- MPOYJPINNSIHAK-UHFFFAOYSA-N cyprodenate Chemical compound CN(C)CCOC(=O)CCC1CCCCC1 MPOYJPINNSIHAK-UHFFFAOYSA-N 0.000 description 1
- 229960004133 cyprodenate Drugs 0.000 description 1
- 229960001140 cyproheptadine Drugs 0.000 description 1
- JJCFRYNCJDLXIK-UHFFFAOYSA-N cyproheptadine Chemical compound C1CN(C)CCC1=C1C2=CC=CC=C2C=CC2=CC=CC=C21 JJCFRYNCJDLXIK-UHFFFAOYSA-N 0.000 description 1
- 229950010806 cyprolidol Drugs 0.000 description 1
- UWFYSQMTEOIJJG-FDTZYFLXSA-N cyproterone acetate Chemical compound C1=C(Cl)C2=CC(=O)[C@@H]3C[C@@H]3[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 UWFYSQMTEOIJJG-FDTZYFLXSA-N 0.000 description 1
- 229960000978 cyproterone acetate Drugs 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- KWGRBVOPPLSCSI-UHFFFAOYSA-N d-ephedrine Natural products CNC(C)C(O)C1=CC=CC=C1 KWGRBVOPPLSCSI-UHFFFAOYSA-N 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- HKUCYAHWPVLPFN-UHFFFAOYSA-N dacemazine Chemical compound C1=CC=C2N(C(=O)CN(C)C)C3=CC=CC=C3SC2=C1 HKUCYAHWPVLPFN-UHFFFAOYSA-N 0.000 description 1
- 229950003783 dacemazine Drugs 0.000 description 1
- HSPYGHDTVQJUDE-LURJTMIESA-N dacisteine Chemical compound CC(=O)N[C@H](C(O)=O)CSC(C)=O HSPYGHDTVQJUDE-LURJTMIESA-N 0.000 description 1
- 229950007177 dacisteine Drugs 0.000 description 1
- 229950004323 dacuronium bromide Drugs 0.000 description 1
- 229950002311 dagapamil Drugs 0.000 description 1
- 229950010011 dalbraminol Drugs 0.000 description 1
- 229950009245 daledalin Drugs 0.000 description 1
- YFAIJBZEDDOCAN-UHFFFAOYSA-N daledalin Chemical compound C12=CC=CC=C2C(CCCNC)(C)CN1C1=CC=CC=C1 YFAIJBZEDDOCAN-UHFFFAOYSA-N 0.000 description 1
- 229950008150 daltroban Drugs 0.000 description 1
- 229950010036 dametralast Drugs 0.000 description 1
- 229950004827 damotepine Drugs 0.000 description 1
- 229960000766 danazol Drugs 0.000 description 1
- POZRVZJJTULAOH-LHZXLZLDSA-N danazol Chemical compound C1[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=CC2=C1C=NO2 POZRVZJJTULAOH-LHZXLZLDSA-N 0.000 description 1
- 229950008019 danitracen Drugs 0.000 description 1
- 229950002395 danosteine Drugs 0.000 description 1
- 108010010449 dansyl-glutamyl-glycyl-arginyl-factor VIIa Proteins 0.000 description 1
- 229960001987 dantrolene Drugs 0.000 description 1
- 229960001577 dantron Drugs 0.000 description 1
- RFWZESUMWJKKRN-UHFFFAOYSA-N dapiprazole Chemical compound CC1=CC=CC=C1N1CCN(CCC=2N3CCCCC3=NN=2)CC1 RFWZESUMWJKKRN-UHFFFAOYSA-N 0.000 description 1
- 229960002947 dapiprazole Drugs 0.000 description 1
- DOAKLVKFURWEDJ-QCMAZARJSA-N daptomycin Chemical compound C([C@H]1C(=O)O[C@H](C)[C@@H](C(NCC(=O)N[C@@H](CCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)N[C@H](CO)C(=O)N[C@H](C(=O)N1)[C@H](C)CC(O)=O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](CC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)CCCCCCCCC)C(=O)C1=CC=CC=C1N DOAKLVKFURWEDJ-QCMAZARJSA-N 0.000 description 1
- 229960005484 daptomycin Drugs 0.000 description 1
- 229950001799 darenzepine Drugs 0.000 description 1
- 229950009702 darodipine Drugs 0.000 description 1
- QERUYFVNIOLCHV-UHFFFAOYSA-N darodipine Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OCC)C1C1=CC=CC2=NON=C12 QERUYFVNIOLCHV-UHFFFAOYSA-N 0.000 description 1
- 229950004062 datelliptium chloride Drugs 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229950010693 dazadrol Drugs 0.000 description 1
- 229950005551 dazepinil Drugs 0.000 description 1
- 229950011349 dazidamine Drugs 0.000 description 1
- 229950002731 dazmegrel Drugs 0.000 description 1
- 229950001971 dazolicine Drugs 0.000 description 1
- YFXIKEZOBJFVAQ-UHFFFAOYSA-N dazopride Chemical compound C1N(CC)N(CC)CC1NC(=O)C1=CC(Cl)=C(N)C=C1OC YFXIKEZOBJFVAQ-UHFFFAOYSA-N 0.000 description 1
- 229950005815 dazopride Drugs 0.000 description 1
- 229950010324 dazoquinast Drugs 0.000 description 1
- OBATZBGFDSVCJD-UHFFFAOYSA-N de-O-acetyl-lanatoside C Natural products CC1OC(OC2CC3C(C4C(C5(CCC(C5(C)C(O)C4)C=4COC(=O)C=4)O)CC3)(C)CC2)CC(O)C1OC(OC1C)CC(O)C1OC(OC1C)CC(O)C1OC1OC(CO)C(O)C(O)C1O OBATZBGFDSVCJD-UHFFFAOYSA-N 0.000 description 1
- 108700027018 deaminooxytocin Proteins 0.000 description 1
- 229950006875 deanol aceglumate Drugs 0.000 description 1
- 229950000059 deboxamet Drugs 0.000 description 1
- JWPGJSVJDAJRLW-UHFFFAOYSA-N debrisoquin Chemical compound C1=CC=C2CN(C(=N)N)CCC2=C1 JWPGJSVJDAJRLW-UHFFFAOYSA-N 0.000 description 1
- 229960004096 debrisoquine Drugs 0.000 description 1
- 229950000405 decamethonium Drugs 0.000 description 1
- BELUJIRQWUXEKA-UHFFFAOYSA-N decanedioic acid;octanedioic acid Chemical compound OC(=O)CCCCCCC(O)=O.OC(=O)CCCCCCCCC(O)=O BELUJIRQWUXEKA-UHFFFAOYSA-N 0.000 description 1
- REYUZOLYIOQRIG-UHFFFAOYSA-N decimemide Chemical compound CCCCCCCCCCOC1=C(OC)C=C(C(N)=O)C=C1OC REYUZOLYIOQRIG-UHFFFAOYSA-N 0.000 description 1
- 229950011023 decimemide Drugs 0.000 description 1
- 229950011368 decitropine Drugs 0.000 description 1
- 229950004366 declenperone Drugs 0.000 description 1
- 229950002790 decloxizine Drugs 0.000 description 1
- 229950005681 decominol Drugs 0.000 description 1
- 229960001878 decoquinate Drugs 0.000 description 1
- JHAYEQICABJSTP-UHFFFAOYSA-N decoquinate Chemical compound N1C=C(C(=O)OCC)C(=O)C2=C1C=C(OCC)C(OCCCCCCCCCC)=C2 JHAYEQICABJSTP-UHFFFAOYSA-N 0.000 description 1
- 229950011040 deditonium bromide Drugs 0.000 description 1
- 229960000958 deferoxamine Drugs 0.000 description 1
- 229960001145 deflazacort Drugs 0.000 description 1
- FBHSPRKOSMHSIF-GRMWVWQJSA-N deflazacort Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(C)=N[C@@]3(C(=O)COC(=O)C)[C@@]1(C)C[C@@H]2O FBHSPRKOSMHSIF-GRMWVWQJSA-N 0.000 description 1
- 229950004239 defosfamide Drugs 0.000 description 1
- 229940061632 dehydroacetic acid Drugs 0.000 description 1
- 235000019258 dehydroacetic acid Nutrition 0.000 description 1
- JEQRBTDTEKWZBW-UHFFFAOYSA-N dehydroacetic acid Chemical compound CC(=O)C1=C(O)OC(C)=CC1=O JEQRBTDTEKWZBW-UHFFFAOYSA-N 0.000 description 1
- PGRHXDWITVMQBC-UHFFFAOYSA-N dehydroacetic acid Natural products CC(=O)C1C(=O)OC(C)=CC1=O PGRHXDWITVMQBC-UHFFFAOYSA-N 0.000 description 1
- XXLZPUYGHQWHRN-RPBOFIJWSA-N dehydroemetine Chemical compound COC1=C(OC)C=C2[C@@H]3CC(C[C@@H]4C5=CC(OC)=C(OC)C=C5CCN4)=C(CC)CN3CCC2=C1 XXLZPUYGHQWHRN-RPBOFIJWSA-N 0.000 description 1
- GDONNNQFENTLQC-VWTPSIDOSA-N delanterone Chemical compound C1C[C@@H]2[C@@]3(C)[C@@H](C)CC(=O)C=C3CC[C@H]2[C@@H]2CC=C[C@]21C GDONNNQFENTLQC-VWTPSIDOSA-N 0.000 description 1
- 229950004137 delanterone Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- 229950006212 delergotrile Drugs 0.000 description 1
- 229950010199 delfantrine Drugs 0.000 description 1
- CGBCCZZJVKUAMX-DFXBJWIESA-N delmadinone acetate Chemical compound C1=C(Cl)C2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 CGBCCZZJVKUAMX-DFXBJWIESA-N 0.000 description 1
- 229950006075 delmadinone acetate Drugs 0.000 description 1
- 229950001116 delmetacin Drugs 0.000 description 1
- QSFOWAYMMZCQNF-UHFFFAOYSA-N delmopinol Chemical compound CCCC(CCC)CCCC1COCCN1CCO QSFOWAYMMZCQNF-UHFFFAOYSA-N 0.000 description 1
- 229960003854 delmopinol Drugs 0.000 description 1
- CHIFCDOIPRCHCF-UHFFFAOYSA-N delorazepam Chemical compound C12=CC(Cl)=CC=C2NC(=O)CN=C1C1=CC=CC=C1Cl CHIFCDOIPRCHCF-UHFFFAOYSA-N 0.000 description 1
- 229950007393 delorazepam Drugs 0.000 description 1
- 229950007806 delprostenate Drugs 0.000 description 1
- CYQFCXCEBYINGO-IAGOWNOFSA-N delta1-THC Chemical compound C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@@H]21 CYQFCXCEBYINGO-IAGOWNOFSA-N 0.000 description 1
- 229950006699 dembrexine Drugs 0.000 description 1
- 229960003715 demecarium bromide Drugs 0.000 description 1
- YHKBUDZECQDYBR-UHFFFAOYSA-L demecarium bromide Chemical compound [Br-].[Br-].C=1C=CC([N+](C)(C)C)=CC=1OC(=O)N(C)CCCCCCCCCCN(C)C(=O)OC1=CC=CC([N+](C)(C)C)=C1 YHKBUDZECQDYBR-UHFFFAOYSA-L 0.000 description 1
- 229960002398 demeclocycline Drugs 0.000 description 1
- 229960005052 demecolcine Drugs 0.000 description 1
- 229950007920 demecycline Drugs 0.000 description 1
- JWAHBTQSSMYISL-MHTWAQMVSA-N demegestone Chemical compound C1CC2=CC(=O)CCC2=C2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(C)[C@@]1(C)CC2 JWAHBTQSSMYISL-MHTWAQMVSA-N 0.000 description 1
- 229960001853 demegestone Drugs 0.000 description 1
- 229950011327 demelverine Drugs 0.000 description 1
- JCSGAUKCDAVARS-UHFFFAOYSA-N demethyltetracycline Natural products CN(C1C(=C(C(C2(C(=C3C(C4=C(C=CC=C4C(C3CC12)O)O)=O)O)O)=O)C(=O)N)O)C JCSGAUKCDAVARS-UHFFFAOYSA-N 0.000 description 1
- 229950010189 demexiptiline Drugs 0.000 description 1
- SEDQWOMFMIJKCU-UHFFFAOYSA-N demexiptiline Chemical compound C1=CC2=CC=CC=C2C(=NOCCNC)C2=CC=CC=C21 SEDQWOMFMIJKCU-UHFFFAOYSA-N 0.000 description 1
- 229950004060 democonazole Drugs 0.000 description 1
- GGRWZBVSUZZMKS-UHFFFAOYSA-N demoxepam Chemical compound C12=CC(Cl)=CC=C2NC(=O)C[N+]([O-])=C1C1=CC=CC=C1 GGRWZBVSUZZMKS-UHFFFAOYSA-N 0.000 description 1
- 229950010734 demoxepam Drugs 0.000 description 1
- FPTOUQZVCUIPHY-UHFFFAOYSA-N denaverine Chemical compound C=1C=CC=CC=1C(C(=O)OCCN(C)C)(OCC(CC)CC)C1=CC=CC=C1 FPTOUQZVCUIPHY-UHFFFAOYSA-N 0.000 description 1
- 229960003392 denaverine Drugs 0.000 description 1
- 229950004687 denbufylline Drugs 0.000 description 1
- 229950001774 denipride Drugs 0.000 description 1
- VHSBBVZJABQOSG-MRXNPFEDSA-N denopamine Chemical compound C1=C(OC)C(OC)=CC=C1CCNC[C@@H](O)C1=CC=C(O)C=C1 VHSBBVZJABQOSG-MRXNPFEDSA-N 0.000 description 1
- 229950007304 denopamine Drugs 0.000 description 1
- 229950007549 denpidazone Drugs 0.000 description 1
- 229950008810 denzimol Drugs 0.000 description 1
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 229950007233 depramine Drugs 0.000 description 1
- AFBYHZACPPSJKD-UHFFFAOYSA-N depramine Chemical compound C1=CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 AFBYHZACPPSJKD-UHFFFAOYSA-N 0.000 description 1
- 229950007161 deprodone Drugs 0.000 description 1
- 229950004449 deprostil Drugs 0.000 description 1
- 229960004073 deptropine Drugs 0.000 description 1
- ZWPODSUQWXAZNC-PMOLBWCYSA-N deptropine Chemical compound C12=CC=CC=C2CCC2=CC=CC=C2C1O[C@H](C1)C[C@H]2CC[C@@H]1N2C ZWPODSUQWXAZNC-PMOLBWCYSA-N 0.000 description 1
- 229950011631 derpanicate Drugs 0.000 description 1
- 229960003800 desaspidin Drugs 0.000 description 1
- 229960001993 deserpidine Drugs 0.000 description 1
- ISMCNVNDWFIXLM-WCGOZPBSSA-N deserpidine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C([C]5C=CC=CC5=N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 ISMCNVNDWFIXLM-WCGOZPBSSA-N 0.000 description 1
- 229960003914 desipramine Drugs 0.000 description 1
- OBATZBGFDSVCJD-LALPQLPRSA-N deslanoside Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@@H]1C[C@@H]2[C@]([C@@H]3[C@H]([C@]4(CC[C@@H]([C@@]4(C)[C@H](O)C3)C=3COC(=O)C=3)O)CC2)(C)CC1)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O OBATZBGFDSVCJD-LALPQLPRSA-N 0.000 description 1
- 229960001324 deslanoside Drugs 0.000 description 1
- JRPANCYSRUEJDY-UHFFFAOYSA-N desmethylmoramide Chemical compound C1COCCN1CCC(C=1C=CC=CC=1)(C=1C=CC=CC=1)C(=O)N1CCCC1 JRPANCYSRUEJDY-UHFFFAOYSA-N 0.000 description 1
- 229950005406 desmethylmoramide Drugs 0.000 description 1
- 229950007334 desocriptine Drugs 0.000 description 1
- OPAVODIYXVLOOM-XAOZLXPPSA-N desocriptine Chemical compound C1=CC([C@H]2C[C@H](CN(C)[C@H]2C2)C(=O)N[C@]3(C(C)C)O[C@@]4(O)[C@@H]5CCCN5C[C@@H](N4C3=O)[C@@H](C)CC)=C3C2=CNC3=C1 OPAVODIYXVLOOM-XAOZLXPPSA-N 0.000 description 1
- RPLCPCMSCLEKRS-BPIQYHPVSA-N desogestrel Chemical compound C1CC[C@@H]2[C@H]3C(=C)C[C@](CC)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 RPLCPCMSCLEKRS-BPIQYHPVSA-N 0.000 description 1
- 229960004976 desogestrel Drugs 0.000 description 1
- 229950003851 desomorphine Drugs 0.000 description 1
- LNNWVNGFPYWNQE-GMIGKAJZSA-N desomorphine Chemical compound C1C2=CC=C(O)C3=C2[C@]24CCN(C)[C@H]1[C@@H]2CCC[C@@H]4O3 LNNWVNGFPYWNQE-GMIGKAJZSA-N 0.000 description 1
- WBGKWQHBNHJJPZ-LECWWXJVSA-N desonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O WBGKWQHBNHJJPZ-LECWWXJVSA-N 0.000 description 1
- 229960003662 desonide Drugs 0.000 description 1
- 229960002593 desoximetasone Drugs 0.000 description 1
- VWVSBHGCDBMOOT-IIEHVVJPSA-N desoximetasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@H](C(=O)CO)[C@@]1(C)C[C@@H]2O VWVSBHGCDBMOOT-IIEHVVJPSA-N 0.000 description 1
- 229960004486 desoxycorticosterone acetate Drugs 0.000 description 1
- 229950008390 desoxycorticosterone pivalate Drugs 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 229960004547 detajmium bitartrate Drugs 0.000 description 1
- 229950004214 detanosal Drugs 0.000 description 1
- 229950010161 deterenol Drugs 0.000 description 1
- JXMXDKHEZLKQPB-UHFFFAOYSA-N detomidine Chemical compound CC1=CC=CC(CC=2[N]C=NC=2)=C1C JXMXDKHEZLKQPB-UHFFFAOYSA-N 0.000 description 1
- 229960001894 detomidine Drugs 0.000 description 1
- 229950003913 detorubicin Drugs 0.000 description 1
- 229950007279 devapamil Drugs 0.000 description 1
- VMVKIDPOEOLUFS-UHFFFAOYSA-N devapamil Chemical compound COC1=CC=CC(CCN(C)CCCC(C#N)(C(C)C)C=2C=C(OC)C(OC)=CC=2)=C1 VMVKIDPOEOLUFS-UHFFFAOYSA-N 0.000 description 1
- 229960003657 dexamethasone acetate Drugs 0.000 description 1
- CIWBQSYVNNPZIQ-PKWREOPISA-N dexamethasone dipropionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CIWBQSYVNNPZIQ-PKWREOPISA-N 0.000 description 1
- 229950000250 dexamethasone dipropionate Drugs 0.000 description 1
- VQODGRNSFPNSQE-CXSFZGCWSA-N dexamethasone phosphate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)COP(O)(O)=O)(O)[C@@]1(C)C[C@@H]2O VQODGRNSFPNSQE-CXSFZGCWSA-N 0.000 description 1
- 229960004833 dexamethasone phosphate Drugs 0.000 description 1
- 229960000632 dexamfetamine Drugs 0.000 description 1
- 229950002495 dexamisole Drugs 0.000 description 1
- ZDIGNSYAACHWNL-HNNXBMFYSA-N dexbrompheniramine Chemical compound C1([C@H](CCN(C)C)C=2N=CC=CC=2)=CC=C(Br)C=C1 ZDIGNSYAACHWNL-HNNXBMFYSA-N 0.000 description 1
- 229960002691 dexbrompheniramine Drugs 0.000 description 1
- SOYKEARSMXGVTM-HNNXBMFYSA-N dexchlorpheniramine Chemical compound C1([C@H](CCN(C)C)C=2N=CC=CC=2)=CC=C(Cl)C=C1 SOYKEARSMXGVTM-HNNXBMFYSA-N 0.000 description 1
- 229960001882 dexchlorpheniramine Drugs 0.000 description 1
- UPMOVJBGNREKJV-CQOQZXRMSA-N dexclamol Chemical compound C12=CC=CC=C2CCC2=CC=CC3=C2[C@@H]1CN1CC[C@@](C(C)C)(O)C[C@@H]13 UPMOVJBGNREKJV-CQOQZXRMSA-N 0.000 description 1
- 229950005215 dexclamol Drugs 0.000 description 1
- LQQIVYSCPWCSSD-HSZRJFAPSA-N dexetimide Chemical compound O=C1NC(=O)CC[C@@]1(C=1C=CC=CC=1)C1CCN(CC=2C=CC=CC=2)CC1 LQQIVYSCPWCSSD-HSZRJFAPSA-N 0.000 description 1
- 229960001908 dexetimide Drugs 0.000 description 1
- 229960004597 dexfenfluramine Drugs 0.000 description 1
- 229950004216 deximafen Drugs 0.000 description 1
- 229950007331 dexindoprofen Drugs 0.000 description 1
- 229950003099 dexivacaine Drugs 0.000 description 1
- 229950001625 dexlofexidine Drugs 0.000 description 1
- 229960004253 dexmedetomidine Drugs 0.000 description 1
- HRLIOXLXPOHXTA-NSHDSACASA-N dexmedetomidine Chemical compound C1([C@@H](C)C=2C(=C(C)C=CC=2)C)=CN=C[N]1 HRLIOXLXPOHXTA-NSHDSACASA-N 0.000 description 1
- 229950004665 dexoxadrol Drugs 0.000 description 1
- 229960003949 dexpanthenol Drugs 0.000 description 1
- 229950008954 dexpropranolol Drugs 0.000 description 1
- 229950005580 dexproxibutene Drugs 0.000 description 1
- WDEFBBTXULIOBB-WBVHZDCISA-N dextilidine Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)OCC)CCC=C[C@H]1N(C)C WDEFBBTXULIOBB-WBVHZDCISA-N 0.000 description 1
- 229950006552 dextilidine Drugs 0.000 description 1
- 229950011181 dextrofemine Drugs 0.000 description 1
- MKXZASYAUGDDCJ-NJAFHUGGSA-N dextromethorphan Chemical compound C([C@@H]12)CCC[C@]11CCN(C)[C@H]2CC2=CC=C(OC)C=C21 MKXZASYAUGDDCJ-NJAFHUGGSA-N 0.000 description 1
- 229960001985 dextromethorphan Drugs 0.000 description 1
- 229960003701 dextromoramide Drugs 0.000 description 1
- INUNXTSAACVKJS-OAQYLSRUSA-N dextromoramide Chemical compound C([C@@H](C)C(C(=O)N1CCCC1)(C=1C=CC=CC=1)C=1C=CC=CC=1)N1CCOCC1 INUNXTSAACVKJS-OAQYLSRUSA-N 0.000 description 1
- JAQUASYNZVUNQP-PVAVHDDUSA-N dextrorphan Chemical compound C1C2=CC=C(O)C=C2[C@@]23CCN(C)[C@@H]1[C@H]2CCCC3 JAQUASYNZVUNQP-PVAVHDDUSA-N 0.000 description 1
- 229950006878 dextrorphan Drugs 0.000 description 1
- 229960001767 dextrothyroxine Drugs 0.000 description 1
- 229950010621 dezaguanine Drugs 0.000 description 1
- 229960003461 dezocine Drugs 0.000 description 1
- VTMVHDZWSFQSQP-VBNZEHGJSA-N dezocine Chemical compound C1CCCC[C@H]2CC3=CC=C(O)C=C3[C@]1(C)[C@H]2N VTMVHDZWSFQSQP-VBNZEHGJSA-N 0.000 description 1
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 1
- 239000012969 di-tertiary-butyl peroxide Substances 0.000 description 1
- 229960004590 diacerein Drugs 0.000 description 1
- 229950004220 diacetamate Drugs 0.000 description 1
- AWOGXJOBNAWQSF-UHFFFAOYSA-N diacetolol Chemical compound CC(C)NCC(O)COC1=CC=C(NC(C)=O)C=C1C(C)=O AWOGXJOBNAWQSF-UHFFFAOYSA-N 0.000 description 1
- 229950003563 diacetolol Drugs 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- 229950010198 diamfenetide Drugs 0.000 description 1
- 125000004420 diamide group Chemical group 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- 229950005813 diamocaine Drugs 0.000 description 1
- 229960002069 diamorphine Drugs 0.000 description 1
- RXTHKWVSXOIHJS-UHFFFAOYSA-N diampromide Chemical compound C=1C=CC=CC=1N(C(=O)CC)CC(C)N(C)CCC1=CC=CC=C1 RXTHKWVSXOIHJS-UHFFFAOYSA-N 0.000 description 1
- 229950001059 diampromide Drugs 0.000 description 1
- 229950000758 dianhydrogalactitol Drugs 0.000 description 1
- 229950000023 diarbarone Drugs 0.000 description 1
- 229950002043 diathymosulfone Drugs 0.000 description 1
- LDBTVAXGKYIFHO-UHFFFAOYSA-N diaveridine Chemical compound C1=C(OC)C(OC)=CC=C1CC1=CN=C(N)N=C1N LDBTVAXGKYIFHO-UHFFFAOYSA-N 0.000 description 1
- 229950000246 diaveridine Drugs 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- FHIVAFMUCKRCQO-UHFFFAOYSA-N diazinon Chemical compound CCOP(=S)(OCC)OC1=CC(C)=NC(C(C)C)=N1 FHIVAFMUCKRCQO-UHFFFAOYSA-N 0.000 description 1
- WVYXNIXAMZOZFK-UHFFFAOYSA-N diaziquone Chemical compound O=C1C(NC(=O)OCC)=C(N2CC2)C(=O)C(NC(=O)OCC)=C1N1CC1 WVYXNIXAMZOZFK-UHFFFAOYSA-N 0.000 description 1
- 229950002389 diaziquone Drugs 0.000 description 1
- 150000008049 diazo compounds Chemical group 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- 150000001989 diazonium salts Chemical class 0.000 description 1
- 229960004042 diazoxide Drugs 0.000 description 1
- 229960003807 dibekacin Drugs 0.000 description 1
- JJCQSGDBDPYCEO-XVZSLQNASA-N dibekacin Chemical compound O1[C@H](CN)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N JJCQSGDBDPYCEO-XVZSLQNASA-N 0.000 description 1
- 229950008416 dibemethine Drugs 0.000 description 1
- 229960003075 dibenzepin Drugs 0.000 description 1
- 229960000493 dibrompropamidine Drugs 0.000 description 1
- GMJFVGRUYJHMCO-UHFFFAOYSA-N dibrompropamidine Chemical compound BrC1=CC(C(=N)N)=CC=C1OCCCOC1=CC=C(C(N)=N)C=C1Br GMJFVGRUYJHMCO-UHFFFAOYSA-N 0.000 description 1
- 229950005821 dibromsalan Drugs 0.000 description 1
- 229950007457 dibrospidium chloride Drugs 0.000 description 1
- 229950010414 dibuprol Drugs 0.000 description 1
- 229950009887 dibupyrone Drugs 0.000 description 1
- 229950000016 dibusadol Drugs 0.000 description 1
- 229950010754 dicarbine Drugs 0.000 description 1
- 229950008815 dicarfen Drugs 0.000 description 1
- 229950009888 dichlorisone Drugs 0.000 description 1
- 229950003772 dichlormezanone Drugs 0.000 description 1
- VJVMIIAKISKTDK-UHFFFAOYSA-N dichloro-[4-[dichloro(methyl)silyl]butyl]-methylsilane Chemical compound C[Si](Cl)(Cl)CCCC[Si](C)(Cl)Cl VJVMIIAKISKTDK-UHFFFAOYSA-N 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- UMNKXPULIDJLSU-UHFFFAOYSA-N dichlorofluoromethane Chemical compound FC(Cl)Cl UMNKXPULIDJLSU-UHFFFAOYSA-N 0.000 description 1
- 229960003887 dichlorophen Drugs 0.000 description 1
- GCFPPDBNEBHYGT-UHFFFAOYSA-N dichlorophenarsine Chemical compound NC1=CC([As](Cl)Cl)=CC=C1O GCFPPDBNEBHYGT-UHFFFAOYSA-N 0.000 description 1
- 229950006869 dichlorophenarsine Drugs 0.000 description 1
- 229950010569 dichloroxylenol Drugs 0.000 description 1
- OEBRKCOSUFCWJD-UHFFFAOYSA-N dichlorvos Chemical compound COP(=O)(OC)OC=C(Cl)Cl OEBRKCOSUFCWJD-UHFFFAOYSA-N 0.000 description 1
- 229950001327 dichlorvos Drugs 0.000 description 1
- 229950010326 diciferron Drugs 0.000 description 1
- 229950001803 dicirenone Drugs 0.000 description 1
- 229960000248 diclazuril Drugs 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- 229960005081 diclofenamide Drugs 0.000 description 1
- GJQPMPFPNINLKP-UHFFFAOYSA-N diclofenamide Chemical compound NS(=O)(=O)C1=CC(Cl)=C(Cl)C(S(N)(=O)=O)=C1 GJQPMPFPNINLKP-UHFFFAOYSA-N 0.000 description 1
- ZJDCGVDEEHWEIG-UHFFFAOYSA-N diclofensine Chemical compound C1N(C)CC2=CC(OC)=CC=C2C1C1=CC=C(Cl)C(Cl)=C1 ZJDCGVDEEHWEIG-UHFFFAOYSA-N 0.000 description 1
- 229950007329 diclofensine Drugs 0.000 description 1
- 229950005189 diclofurime Drugs 0.000 description 1
- 229950004357 diclometide Drugs 0.000 description 1
- 229950000233 diclonixin Drugs 0.000 description 1
- 229960004917 dicobalt edetate Drugs 0.000 description 1
- 229950008863 dicolinium iodide Drugs 0.000 description 1
- DOBMPNYZJYQDGZ-UHFFFAOYSA-N dicoumarol Chemical compound C1=CC=CC2=C1OC(=O)C(CC=1C(OC3=CC=CC=C3C=1O)=O)=C2O DOBMPNYZJYQDGZ-UHFFFAOYSA-N 0.000 description 1
- 229960001912 dicoumarol Drugs 0.000 description 1
- 229950006317 dicresulene Drugs 0.000 description 1
- HIZKPJUTKKJDGA-UHFFFAOYSA-N dicumarol Natural products O=C1OC2=CC=CC=C2C(=O)C1CC1C(=O)C2=CC=CC=C2OC1=O HIZKPJUTKKJDGA-UHFFFAOYSA-N 0.000 description 1
- CURUTKGFNZGFSE-UHFFFAOYSA-N dicyclomine Chemical compound C1CCCCC1C1(C(=O)OCCN(CC)CC)CCCCC1 CURUTKGFNZGFSE-UHFFFAOYSA-N 0.000 description 1
- 229960002777 dicycloverine Drugs 0.000 description 1
- 229930189582 didemnin Natural products 0.000 description 1
- 229950005934 didrovaltrate Drugs 0.000 description 1
- PVKOJQHHDYMUEI-UHFFFAOYSA-N didrovaltrate Natural products CC(C)CC(=O)COC1=COC(OC(=O)CC(C)C)C2C1CC(OC(=O)C)C23CO3 PVKOJQHHDYMUEI-UHFFFAOYSA-N 0.000 description 1
- 229950006824 dieldrin Drugs 0.000 description 1
- DFBKLUNHFCTMDC-PICURKEMSA-N dieldrin Chemical compound C([C@H]1[C@H]2[C@@]3(Cl)C(Cl)=C([C@]([C@H]22)(Cl)C3(Cl)Cl)Cl)[C@H]2[C@@H]2[C@H]1O2 DFBKLUNHFCTMDC-PICURKEMSA-N 0.000 description 1
- NGPMUTDCEIKKFM-UHFFFAOYSA-N dieldrin Natural products CC1=C(Cl)C2(Cl)C3C4CC(C5OC45)C3C1(Cl)C2(Cl)Cl NGPMUTDCEIKKFM-UHFFFAOYSA-N 0.000 description 1
- 229960003839 dienestrol Drugs 0.000 description 1
- NFDFQCUYFHCNBW-SCGPFSFSSA-N dienestrol Chemical compound C=1C=C(O)C=CC=1\C(=C/C)\C(=C\C)\C1=CC=C(O)C=C1 NFDFQCUYFHCNBW-SCGPFSFSSA-N 0.000 description 1
- AZFLJNIPTRTECV-FUMNGEBKSA-N dienogest Chemical compound C1CC(=O)C=C2CC[C@@H]([C@H]3[C@@](C)([C@](CC3)(O)CC#N)CC3)C3=C21 AZFLJNIPTRTECV-FUMNGEBKSA-N 0.000 description 1
- 229960003309 dienogest Drugs 0.000 description 1
- ORTYMGHCFWKXHO-UHFFFAOYSA-N diethadione Chemical compound CCC1(CC)COC(=O)NC1=O ORTYMGHCFWKXHO-UHFFFAOYSA-N 0.000 description 1
- 229960003675 diethadione Drugs 0.000 description 1
- LURMIKVZJSMXQE-UHFFFAOYSA-N diethazine Chemical compound C1=CC=C2N(CCN(CC)CC)C3=CC=CC=C3SC2=C1 LURMIKVZJSMXQE-UHFFFAOYSA-N 0.000 description 1
- 229950005520 diethazine Drugs 0.000 description 1
- LMBWSYZSUOEYSN-UHFFFAOYSA-N diethyldithiocarbamic acid Chemical compound CCN(CC)C(S)=S LMBWSYZSUOEYSN-UHFFFAOYSA-N 0.000 description 1
- XXEPPPIWZFICOJ-UHFFFAOYSA-N diethylpropion Chemical compound CCN(CC)C(C)C(=O)C1=CC=CC=C1 XXEPPPIWZFICOJ-UHFFFAOYSA-N 0.000 description 1
- 229960004890 diethylpropion Drugs 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- NLORYLAYLIXTID-ISLYRVAYSA-N diethylstilbestrol diphosphate Chemical compound C=1C=C(OP(O)(O)=O)C=CC=1C(/CC)=C(\CC)C1=CC=C(OP(O)(O)=O)C=C1 NLORYLAYLIXTID-ISLYRVAYSA-N 0.000 description 1
- CBYWMRHUUVRIAF-UHFFFAOYSA-N diethylthiambutene Chemical compound C=1C=CSC=1C(=CC(C)N(CC)CC)C1=CC=CS1 CBYWMRHUUVRIAF-UHFFFAOYSA-N 0.000 description 1
- 229950009987 diethylthiambutene Drugs 0.000 description 1
- 229960001673 diethyltoluamide Drugs 0.000 description 1
- 229950007672 dietifen Drugs 0.000 description 1
- GJJRIOLBUILIGK-UHFFFAOYSA-N difebarbamate Chemical compound O=C1N(CC(COCCCC)OC(N)=O)C(=O)N(CC(COCCCC)OC(N)=O)C(=O)C1(CC)C1=CC=CC=C1 GJJRIOLBUILIGK-UHFFFAOYSA-N 0.000 description 1
- 229960000694 difebarbamate Drugs 0.000 description 1
- 229960004944 difemerine Drugs 0.000 description 1
- WJIZVQNUJVMJAZ-UHFFFAOYSA-N difemerine Chemical compound C=1C=CC=CC=1C(O)(C(=O)OC(C)(C)CN(C)C)C1=CC=CC=C1 WJIZVQNUJVMJAZ-UHFFFAOYSA-N 0.000 description 1
- UHZJPVXMMCFPNR-UHFFFAOYSA-N difemetorex Chemical compound OCCN1CCCCC1C(C=1C=CC=CC=1)C1=CC=CC=C1 UHZJPVXMMCFPNR-UHFFFAOYSA-N 0.000 description 1
- 229950011229 difemetorex Drugs 0.000 description 1
- PCXMKBOWWVXEDT-UHFFFAOYSA-N difenamizole Chemical compound CN(C)C(C)C(=O)NC1=CC(C=2C=CC=CC=2)=NN1C1=CC=CC=C1 PCXMKBOWWVXEDT-UHFFFAOYSA-N 0.000 description 1
- 229950000061 difenamizole Drugs 0.000 description 1
- 229950005631 difencloxazine Drugs 0.000 description 1
- 229950001428 difenoximide Drugs 0.000 description 1
- UFIVBRCCIRTJTN-UHFFFAOYSA-N difenoxin Chemical compound C1CC(C(=O)O)(C=2C=CC=CC=2)CCN1CCC(C#N)(C=1C=CC=CC=1)C1=CC=CC=C1 UFIVBRCCIRTJTN-UHFFFAOYSA-N 0.000 description 1
- 229960005493 difenoxin Drugs 0.000 description 1
- YQVALJGIKVYRNI-UHFFFAOYSA-N difetarsone Chemical compound C1=CC([As](O)(=O)O)=CC=C1NCCNC1=CC=C([As](O)(O)=O)C=C1 YQVALJGIKVYRNI-UHFFFAOYSA-N 0.000 description 1
- 229960004941 difetarsone Drugs 0.000 description 1
- 229950001321 difeterol Drugs 0.000 description 1
- 229960002124 diflorasone diacetate Drugs 0.000 description 1
- BOBLHFUVNSFZPJ-JOYXJVLSSA-N diflorasone diacetate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H](C)[C@@](C(=O)COC(C)=O)(OC(C)=O)[C@@]2(C)C[C@@H]1O BOBLHFUVNSFZPJ-JOYXJVLSSA-N 0.000 description 1
- NOCJXYPHIIZEHN-UHFFFAOYSA-N difloxacin Chemical compound C1CN(C)CCN1C(C(=C1)F)=CC2=C1C(=O)C(C(O)=O)=CN2C1=CC=C(F)C=C1 NOCJXYPHIIZEHN-UHFFFAOYSA-N 0.000 description 1
- 229950001733 difloxacin Drugs 0.000 description 1
- 229960004091 diflucortolone Drugs 0.000 description 1
- OGPWIDANBSLJPC-RFPWEZLHSA-N diflucortolone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)CO)[C@@]2(C)C[C@@H]1O OGPWIDANBSLJPC-RFPWEZLHSA-N 0.000 description 1
- 229950001862 diflumidone Drugs 0.000 description 1
- 229960004875 difluprednate Drugs 0.000 description 1
- 229950007956 diftalone Drugs 0.000 description 1
- CKNOLMVLQUPVMU-YMMLYESFSA-N digitalin Chemical compound C1([C@@H]2[C@@]3(C)CC[C@H]4[C@H]([C@]3(C[C@@H]2O)O)CC[C@H]2[C@]4(C)CC[C@@H](C2)O[C@H]2[C@H](O)[C@H]([C@H]([C@@H](C)O2)O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)OC)=CC(=O)OC1 CKNOLMVLQUPVMU-YMMLYESFSA-N 0.000 description 1
- 229950004590 digitalin Drugs 0.000 description 1
- 229960003215 dihexyverine Drugs 0.000 description 1
- MNSQDVCVWNXBFQ-UHFFFAOYSA-N dihexyverine Chemical compound C1CCCCC1(C1CCCCC1)C(=O)OCCN1CCCCC1 MNSQDVCVWNXBFQ-UHFFFAOYSA-N 0.000 description 1
- VQKLRVZQQYVIJW-UHFFFAOYSA-N dihydralazine Chemical compound C1=CC=C2C(NN)=NN=C(NN)C2=C1 VQKLRVZQQYVIJW-UHFFFAOYSA-N 0.000 description 1
- 229960002877 dihydralazine Drugs 0.000 description 1
- 229940069063 dihydro-beta-ergocryptine Drugs 0.000 description 1
- RBOXVHNMENFORY-DNJOTXNNSA-N dihydrocodeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC RBOXVHNMENFORY-DNJOTXNNSA-N 0.000 description 1
- 229960003061 dihydroemetine Drugs 0.000 description 1
- HESHRHUZIWVEAJ-JGRZULCMSA-N dihydroergotamine Chemical compound C([C@H]1C(=O)N2CCC[C@H]2[C@]2(O)O[C@@](C(N21)=O)(C)NC(=O)[C@H]1CN([C@H]2[C@@H](C3=CC=CC4=NC=C([C]34)C2)C1)C)C1=CC=CC=C1 HESHRHUZIWVEAJ-JGRZULCMSA-N 0.000 description 1
- 229960004704 dihydroergotamine Drugs 0.000 description 1
- 229960002222 dihydrostreptomycin Drugs 0.000 description 1
- 229960000465 dihydrotachysterol Drugs 0.000 description 1
- ILYCWAKSDCYMBB-OPCMSESCSA-N dihydrotachysterol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)/C=C/[C@H](C)C(C)C)=C\C=C1/C[C@@H](O)CC[C@@H]1C ILYCWAKSDCYMBB-OPCMSESCSA-N 0.000 description 1
- PHHROXLDZHUIGO-UHFFFAOYSA-N dihydrovaltrate Natural products CC(C)CC(=O)OCC1=COC(OC(=O)CC(C)C)C2C1CC(OC(C)=O)C21CO1 PHHROXLDZHUIGO-UHFFFAOYSA-N 0.000 description 1
- RVIRCOIWERMNCC-UHFFFAOYSA-N diiodomethanesulfonic acid Chemical compound OS(=O)(=O)C(I)I RVIRCOIWERMNCC-UHFFFAOYSA-N 0.000 description 1
- YBJKOPHEJOMRMN-UHFFFAOYSA-N diisopromine Chemical compound C=1C=CC=CC=1C(CCN(C(C)C)C(C)C)C1=CC=CC=C1 YBJKOPHEJOMRMN-UHFFFAOYSA-N 0.000 description 1
- 229960004071 diisopromine Drugs 0.000 description 1
- LVTYICIALWPMFW-UHFFFAOYSA-N diisopropanolamine Chemical compound CC(O)CNCC(C)O LVTYICIALWPMFW-UHFFFAOYSA-N 0.000 description 1
- 229940043276 diisopropanolamine Drugs 0.000 description 1
- 230000000916 dilatatory effect Effects 0.000 description 1
- 229960001079 dilazep Drugs 0.000 description 1
- 229950007942 dilevalol Drugs 0.000 description 1
- 229950011421 dilmefone Drugs 0.000 description 1
- 229960001111 diloxanide Drugs 0.000 description 1
- BDYYDXJSHYEDGB-UHFFFAOYSA-N diloxanide furoate Chemical compound C1=CC(N(C(=O)C(Cl)Cl)C)=CC=C1OC(=O)C1=CC=CO1 BDYYDXJSHYEDGB-UHFFFAOYSA-N 0.000 description 1
- 229950009113 dimabefylline Drugs 0.000 description 1
- NAPSCFZYZVSQHF-UHFFFAOYSA-N dimantine Chemical compound CCCCCCCCCCCCCCCCCCN(C)C NAPSCFZYZVSQHF-UHFFFAOYSA-N 0.000 description 1
- 229950010007 dimantine Drugs 0.000 description 1
- 229960004462 dimazole Drugs 0.000 description 1
- 229950004241 dimecamine Drugs 0.000 description 1
- 229950000045 dimecolonium iodide Drugs 0.000 description 1
- 229960003780 dimecrotic acid Drugs 0.000 description 1
- GAVBHVRHVQMWEI-UHFFFAOYSA-N dimefadane Chemical compound C12=CC=CC=C2C(N(C)C)CC1C1=CC=CC=C1 GAVBHVRHVQMWEI-UHFFFAOYSA-N 0.000 description 1
- 229950010893 dimefadane Drugs 0.000 description 1
- ZXFQRFXLFWWKLX-UHFFFAOYSA-N dimefline Chemical compound CN(C)CC=1C(OC)=CC=C(C(C=2C)=O)C=1OC=2C1=CC=CC=C1 ZXFQRFXLFWWKLX-UHFFFAOYSA-N 0.000 description 1
- 229960000809 dimefline Drugs 0.000 description 1
- 229950007360 dimelazine Drugs 0.000 description 1
- KBEZZLAAKIIPFK-NJAFHUGGSA-N dimemorfan Chemical compound C1C2=CC=C(C)C=C2[C@@]23CCN(C)[C@@H]1[C@H]2CCCC3 KBEZZLAAKIIPFK-NJAFHUGGSA-N 0.000 description 1
- 229960001056 dimemorfan Drugs 0.000 description 1
- MZDOIJOUFRQXHC-UHFFFAOYSA-N dimenhydrinate Chemical compound O=C1N(C)C(=O)N(C)C2=NC(Cl)=N[C]21.C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 MZDOIJOUFRQXHC-UHFFFAOYSA-N 0.000 description 1
- 229960004993 dimenhydrinate Drugs 0.000 description 1
- RHUWRJWFHUKVED-UHFFFAOYSA-N dimenoxadol Chemical compound C=1C=CC=CC=1C(C(=O)OCCN(C)C)(OCC)C1=CC=CC=C1 RHUWRJWFHUKVED-UHFFFAOYSA-N 0.000 description 1
- 229950011187 dimenoxadol Drugs 0.000 description 1
- 229950009815 dimepranol Drugs 0.000 description 1
- 229950003560 dimepregnen Drugs 0.000 description 1
- 229950007232 dimeprozan Drugs 0.000 description 1
- 229960001051 dimercaprol Drugs 0.000 description 1
- WQABCVAJNWAXTE-UHFFFAOYSA-N dimercaprol Chemical compound OCC(S)CS WQABCVAJNWAXTE-UHFFFAOYSA-N 0.000 description 1
- 229950009278 dimesna Drugs 0.000 description 1
- 229950008031 dimesone Drugs 0.000 description 1
- 229960003524 dimetacrine Drugs 0.000 description 1
- RYQOGSFEJBUZBX-UHFFFAOYSA-N dimetacrine Chemical compound C1=CC=C2N(CCCN(C)C)C3=CC=CC=C3C(C)(C)C2=C1 RYQOGSFEJBUZBX-UHFFFAOYSA-N 0.000 description 1
- 229950005104 dimetamfetamine Drugs 0.000 description 1
- 229950004446 dimethadione Drugs 0.000 description 1
- CMKUGKVVUUGBHJ-UHFFFAOYSA-N dimethazan Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2CCN(C)C CMKUGKVVUUGBHJ-UHFFFAOYSA-N 0.000 description 1
- 229950002134 dimethazan Drugs 0.000 description 1
- 229950006690 dimethisterone Drugs 0.000 description 1
- 229950002386 dimetholizine Drugs 0.000 description 1
- 229960004327 dimethoxanate Drugs 0.000 description 1
- OOVJCSPCMCAXEX-UHFFFAOYSA-N dimethoxanate Chemical compound C1=CC=C2N(C(=O)OCCOCCN(C)C)C3=CC=CC=C3SC2=C1 OOVJCSPCMCAXEX-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- UBHZUDXTHNMNLD-UHFFFAOYSA-N dimethylsilane Chemical compound C[SiH2]C UBHZUDXTHNMNLD-UHFFFAOYSA-N 0.000 description 1
- CANBGVXYBPOLRR-UHFFFAOYSA-N dimethylthiambutene Chemical compound C=1C=CSC=1C(=CC(C)N(C)C)C1=CC=CS1 CANBGVXYBPOLRR-UHFFFAOYSA-N 0.000 description 1
- 229950005563 dimethylthiambutene Drugs 0.000 description 1
- IRPSJVWFSWAZSZ-OIUSMDOTSA-L dimethyltubocurarinium chloride Chemical compound [Cl-].[Cl-].C1([C@@H]([N+](CCC1=CC=1OC)(C)C)CC2=CC=C(C=C2)O2)=CC=1OC(=C1)C(OC)=CC=C1C[C@H]1[N+](C)(C)CCC3=C1C2=C(OC)C(OC)=C3 IRPSJVWFSWAZSZ-OIUSMDOTSA-L 0.000 description 1
- 229950003412 dimethyltubocurarinium chloride Drugs 0.000 description 1
- 229960001992 dimetindene Drugs 0.000 description 1
- MVMQESMQSYOVGV-UHFFFAOYSA-N dimetindene Chemical compound CN(C)CCC=1CC2=CC=CC=C2C=1C(C)C1=CC=CC=N1 MVMQESMQSYOVGV-UHFFFAOYSA-N 0.000 description 1
- 229950011369 dimetipirium bromide Drugs 0.000 description 1
- 229960002547 dimetofrine Drugs 0.000 description 1
- ZKGDBJAHIIXDDW-UHFFFAOYSA-N dimetofrine Chemical compound CNCC(O)C1=CC(OC)=C(O)C(OC)=C1 ZKGDBJAHIIXDDW-UHFFFAOYSA-N 0.000 description 1
- VWNWVCJGUMZDIU-UHFFFAOYSA-N dimetotiazine Chemical compound C1=C(S(=O)(=O)N(C)C)C=C2N(CC(C)N(C)C)C3=CC=CC=C3SC2=C1 VWNWVCJGUMZDIU-UHFFFAOYSA-N 0.000 description 1
- 229960001640 dimetotiazine Drugs 0.000 description 1
- IBXPYPUJPLLOIN-UHFFFAOYSA-N dimetridazole Chemical compound CC1=NC=C(N(=O)=O)N1C IBXPYPUJPLLOIN-UHFFFAOYSA-N 0.000 description 1
- 229960000946 dimetridazole Drugs 0.000 description 1
- XNYZHCFCZNMTFY-UHFFFAOYSA-N diminazene Chemical compound C1=CC(C(=N)N)=CC=C1N\N=N\C1=CC=C(C(N)=N)C=C1 XNYZHCFCZNMTFY-UHFFFAOYSA-N 0.000 description 1
- 229950007095 diminazene Drugs 0.000 description 1
- 229950008992 dimoxaprost Drugs 0.000 description 1
- 229950003231 dimpylate Drugs 0.000 description 1
- 229960003724 dimyristoylphosphatidylcholine Drugs 0.000 description 1
- 229950004830 diniprofylline Drugs 0.000 description 1
- 229960003934 dinitolmide Drugs 0.000 description 1
- 229960001342 dinoprost Drugs 0.000 description 1
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 1
- 229960002986 dinoprostone Drugs 0.000 description 1
- 229950007851 dinsed Drugs 0.000 description 1
- 150000002009 diols Chemical group 0.000 description 1
- WXMARHKAXWRNDM-GAMIEDRGSA-N diosgenin 3-O-beta-D-glucoside Chemical compound O([C@@H]1CC2=CC[C@H]3[C@@H]4C[C@H]5[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@@H]([C@]1(OC[C@H](C)CC1)O5)C)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O WXMARHKAXWRNDM-GAMIEDRGSA-N 0.000 description 1
- GZSOSUNBTXMUFQ-YFAPSIMESA-N diosmin Chemical compound C1=C(O)C(OC)=CC=C1C(OC1=C2)=CC(=O)C1=C(O)C=C2O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@H](O)[C@@H](O)[C@H](C)O2)O)O1 GZSOSUNBTXMUFQ-YFAPSIMESA-N 0.000 description 1
- 229960004352 diosmin Drugs 0.000 description 1
- IGBKNLGEMMEWKD-UHFFFAOYSA-N diosmin Natural products COc1ccc(cc1)C2=C(O)C(=O)c3c(O)cc(OC4OC(COC5OC(C)C(O)C(O)C5O)C(O)C(O)C4O)cc3O2 IGBKNLGEMMEWKD-UHFFFAOYSA-N 0.000 description 1
- 229950000186 dioxadilol Drugs 0.000 description 1
- 229950006161 dioxadrol Drugs 0.000 description 1
- 229950001656 dioxamate Drugs 0.000 description 1
- 229950008972 dioxaphetyl butyrate Drugs 0.000 description 1
- LQGIXNQCOXNCRP-UHFFFAOYSA-N dioxaphetyl butyrate Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C(=O)OCC)CCN1CCOCC1 LQGIXNQCOXNCRP-UHFFFAOYSA-N 0.000 description 1
- OHDICGSRVLBVLC-UHFFFAOYSA-N dioxethedrin Chemical compound CCNC(C)C(O)C1=CC=C(O)C(O)=C1 OHDICGSRVLBVLC-UHFFFAOYSA-N 0.000 description 1
- 229950005373 dioxethedrin Drugs 0.000 description 1
- 229950009783 dioxifedrine Drugs 0.000 description 1
- 229960004960 dioxybenzone Drugs 0.000 description 1
- NFABMCCMPXXBFY-UHFFFAOYSA-N dioxyline Chemical compound C1=C(OC)C(OCC)=CC=C1CC1=NC(C)=CC2=CC(OC)=C(OC)C=C12 NFABMCCMPXXBFY-UHFFFAOYSA-N 0.000 description 1
- 229950010935 dioxyline Drugs 0.000 description 1
- 229960002228 diperodon Drugs 0.000 description 1
- 229960000591 diphemanil methylsulfate Drugs 0.000 description 1
- BREMLQBSKCSNNH-UHFFFAOYSA-M diphemanil methylsulfate Chemical compound COS([O-])(=O)=O.C1C[N+](C)(C)CCC1=C(C=1C=CC=CC=1)C1=CC=CC=C1 BREMLQBSKCSNNH-UHFFFAOYSA-M 0.000 description 1
- 229960000267 diphenadione Drugs 0.000 description 1
- ZBJBRUSGEJORQL-UHFFFAOYSA-N diphenan Chemical compound C1=CC(OC(=O)N)=CC=C1CC1=CC=CC=C1 ZBJBRUSGEJORQL-UHFFFAOYSA-N 0.000 description 1
- 229950002048 diphenan Drugs 0.000 description 1
- 229960000520 diphenhydramine Drugs 0.000 description 1
- 229960004192 diphenoxylate Drugs 0.000 description 1
- HYPPXZBJBPSRLK-UHFFFAOYSA-N diphenoxylate Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCC(C#N)(C=1C=CC=CC=1)C1=CC=CC=C1 HYPPXZBJBPSRLK-UHFFFAOYSA-N 0.000 description 1
- ZMISODWVFHHWNR-UHFFFAOYSA-N diphenyl(4-piperidinyl)methanol Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(O)C1CCNCC1 ZMISODWVFHHWNR-UHFFFAOYSA-N 0.000 description 1
- KPUSUIKQQCFQCX-VQTJNVASSA-N diphenyl-[(1r,2r)-2-pyridin-4-ylcyclopropyl]methanol Chemical compound C1([C@@H]2C[C@H]2C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)=CC=NC=C1 KPUSUIKQQCFQCX-VQTJNVASSA-N 0.000 description 1
- 229950011144 diphoxazide Drugs 0.000 description 1
- SVDHSZFEQYXRDC-UHFFFAOYSA-N dipipanone Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C(=O)CC)CC(C)N1CCCCC1 SVDHSZFEQYXRDC-UHFFFAOYSA-N 0.000 description 1
- 229960002500 dipipanone Drugs 0.000 description 1
- WBTCZEPSIIFINA-MSFWTACDSA-J dipotassium;antimony(3+);(2r,3r)-2,3-dioxidobutanedioate;trihydrate Chemical compound O.O.O.[K+].[K+].[Sb+3].[Sb+3].[O-]C(=O)[C@H]([O-])[C@@H]([O-])C([O-])=O.[O-]C(=O)[C@H]([O-])[C@@H]([O-])C([O-])=O WBTCZEPSIIFINA-MSFWTACDSA-J 0.000 description 1
- VDKMYSMWQCFYBQ-UHFFFAOYSA-N diprafenone Chemical compound CCC(C)(C)NCC(O)COC1=CC=CC=C1C(=O)CCC1=CC=CC=C1 VDKMYSMWQCFYBQ-UHFFFAOYSA-N 0.000 description 1
- 229950002948 diprafenone Drugs 0.000 description 1
- 229950002494 diprenorphine Drugs 0.000 description 1
- 229950001668 diprobutine Drugs 0.000 description 1
- 229950008011 diprofene Drugs 0.000 description 1
- 229950001624 diprogulic acid Drugs 0.000 description 1
- 229950000032 diproleandomycin Drugs 0.000 description 1
- 229960002819 diprophylline Drugs 0.000 description 1
- NTGLQWGMESPVBV-UHFFFAOYSA-N diproqualone Chemical compound C1=CC=C2C(=O)N(CC(O)CO)C(C)=NC2=C1 NTGLQWGMESPVBV-UHFFFAOYSA-N 0.000 description 1
- 229950003185 diproqualone Drugs 0.000 description 1
- APMMVXSVJLZZRR-UHFFFAOYSA-N diproteverine Chemical compound C1=C(OCC)C(OCC)=CC=C1CC1=NCCC2=CC(OC(C)C)=C(OC(C)C)C=C12 APMMVXSVJLZZRR-UHFFFAOYSA-N 0.000 description 1
- 229950007849 diproteverine Drugs 0.000 description 1
- 229950004324 diproxadol Drugs 0.000 description 1
- 229960002768 dipyridamole Drugs 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- ZHDBTKPXEJDTTQ-UHFFFAOYSA-N dipyrithione Chemical compound [O-][N+]1=CC=CC=C1SSC1=CC=CC=[N+]1[O-] ZHDBTKPXEJDTTQ-UHFFFAOYSA-N 0.000 description 1
- 229960000842 dipyrocetyl Drugs 0.000 description 1
- 229940120889 dipyrone Drugs 0.000 description 1
- 229960004100 dirithromycin Drugs 0.000 description 1
- WLOHNSSYAXHWNR-NXPDYKKBSA-N dirithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H]2O[C@H](COCCOC)N[C@H]([C@@H]2C)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 WLOHNSSYAXHWNR-NXPDYKKBSA-N 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 229950000781 disobutamide Drugs 0.000 description 1
- YKFWMDHZMQLWBO-UHFFFAOYSA-N disobutamide Chemical compound C=1C=CC=C(Cl)C=1C(CCN(C(C)C)C(C)C)(C(N)=O)CCN1CCCCC1 YKFWMDHZMQLWBO-UHFFFAOYSA-N 0.000 description 1
- VLARUOGDXDTHEH-UHFFFAOYSA-L disodium cromoglycate Chemical compound [Na+].[Na+].O1C(C([O-])=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C([O-])=O)O2 VLARUOGDXDTHEH-UHFFFAOYSA-L 0.000 description 1
- KQYGMURBTJPBPQ-UHFFFAOYSA-L disodium;2-(2-sulfonatoethyldisulfanyl)ethanesulfonate Chemical compound [Na+].[Na+].[O-]S(=O)(=O)CCSSCCS([O-])(=O)=O KQYGMURBTJPBPQ-UHFFFAOYSA-L 0.000 description 1
- 229960004966 disofenin Drugs 0.000 description 1
- UDUSOMRJOPCWHT-UHFFFAOYSA-N disofenin Chemical compound CC(C)C1=CC=CC(C(C)C)=C1NC(=O)CN(CC(O)=O)CC(O)=O UDUSOMRJOPCWHT-UHFFFAOYSA-N 0.000 description 1
- 229950005648 disogluside Drugs 0.000 description 1
- 229960001066 disopyramide Drugs 0.000 description 1
- UVTNFZQICZKOEM-UHFFFAOYSA-N disopyramide Chemical compound C=1C=CC=NC=1C(C(N)=O)(CCN(C(C)C)C(C)C)C1=CC=CC=C1 UVTNFZQICZKOEM-UHFFFAOYSA-N 0.000 description 1
- 229950002098 disoxaril Drugs 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 229960001446 distigmine Drugs 0.000 description 1
- GJHSNEVFXQVOHR-UHFFFAOYSA-L distigmine bromide Chemical compound [Br-].[Br-].C=1C=C[N+](C)=CC=1OC(=O)N(C)CCCCCCN(C)C(=O)OC1=CC=C[N+](C)=C1 GJHSNEVFXQVOHR-UHFFFAOYSA-L 0.000 description 1
- 229950001276 disulergine Drugs 0.000 description 1
- 229950008177 disulfamide Drugs 0.000 description 1
- RCFKEIREOSXLET-UHFFFAOYSA-N disulfamide Chemical compound CC1=CC(Cl)=C(S(N)(=O)=O)C=C1S(N)(=O)=O RCFKEIREOSXLET-UHFFFAOYSA-N 0.000 description 1
- 229960002563 disulfiram Drugs 0.000 description 1
- 229950001169 disuprazole Drugs 0.000 description 1
- 229960005067 ditazole Drugs 0.000 description 1
- UUCMDZWCRNZCOY-UHFFFAOYSA-N ditazole Chemical compound O1C(N(CCO)CCO)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 UUCMDZWCRNZCOY-UHFFFAOYSA-N 0.000 description 1
- 229950008811 ditercalinium chloride Drugs 0.000 description 1
- 229950005765 dithiazanine iodide Drugs 0.000 description 1
- MNQDKWZEUULFPX-UHFFFAOYSA-M dithiazanine iodide Chemical compound [I-].S1C2=CC=CC=C2[N+](CC)=C1C=CC=CC=C1N(CC)C2=CC=CC=C2S1 MNQDKWZEUULFPX-UHFFFAOYSA-M 0.000 description 1
- 229960002311 dithranol Drugs 0.000 description 1
- 229950004394 ditiocarb Drugs 0.000 description 1
- 229950011425 ditiomustine Drugs 0.000 description 1
- 229950001155 ditolamide Drugs 0.000 description 1
- DWGXUDUOJPYAOR-UHFFFAOYSA-N ditophal Chemical compound CCSC(=O)C1=CC=CC(C(=O)SCC)=C1 DWGXUDUOJPYAOR-UHFFFAOYSA-N 0.000 description 1
- 229950006806 ditophal Drugs 0.000 description 1
- 239000002934 diuretic Substances 0.000 description 1
- 229950007948 divabuterol Drugs 0.000 description 1
- 229960002377 dixanthogen Drugs 0.000 description 1
- 229950006278 dizatrifone Drugs 0.000 description 1
- 229950004794 dizocilpine Drugs 0.000 description 1
- LBOJYSIDWZQNJS-CVEARBPZSA-N dizocilpine Chemical compound C12=CC=CC=C2[C@]2(C)C3=CC=CC=C3C[C@H]1N2 LBOJYSIDWZQNJS-CVEARBPZSA-N 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- DLNKOYKMWOXYQA-UHFFFAOYSA-N dl-pseudophenylpropanolamine Natural products CC(N)C(O)C1=CC=CC=C1 DLNKOYKMWOXYQA-UHFFFAOYSA-N 0.000 description 1
- 229950002808 dobupride Drugs 0.000 description 1
- 229960001089 dobutamine Drugs 0.000 description 1
- 229950006045 docarpamine Drugs 0.000 description 1
- 229950000310 doconazole Drugs 0.000 description 1
- 229940018602 docusate Drugs 0.000 description 1
- MVZYGLQQNPFARE-UHFFFAOYSA-N doliracetam Chemical compound C12=CC=CC=C2N(CC(=O)N)C(=O)C1C1=CC=CC=C1 MVZYGLQQNPFARE-UHFFFAOYSA-N 0.000 description 1
- 229950006222 doliracetam Drugs 0.000 description 1
- 229950009799 domazoline Drugs 0.000 description 1
- 229960001700 domiodol Drugs 0.000 description 1
- NEIPZWZQHXCYDV-UHFFFAOYSA-N domiodol Chemical compound OCC1COC(CI)O1 NEIPZWZQHXCYDV-UHFFFAOYSA-N 0.000 description 1
- 229960001859 domiphen bromide Drugs 0.000 description 1
- 229950011469 domipizone Drugs 0.000 description 1
- 229950009486 domoprednate Drugs 0.000 description 1
- IXTXYSAWZICAPV-UHFFFAOYSA-N domoxin Chemical compound C1OC2=CC=CC=C2OC1CN(N)CC1=CC=CC=C1 IXTXYSAWZICAPV-UHFFFAOYSA-N 0.000 description 1
- 229950002967 domoxin Drugs 0.000 description 1
- 229960001253 domperidone Drugs 0.000 description 1
- FGXWKSZFVQUSTL-UHFFFAOYSA-N domperidone Chemical compound C12=CC=CC=C2NC(=O)N1CCCN(CC1)CCC1N1C2=CC=C(Cl)C=C2NC1=O FGXWKSZFVQUSTL-UHFFFAOYSA-N 0.000 description 1
- 229950010956 donetidine Drugs 0.000 description 1
- MURUHMTVTKOWBY-UHFFFAOYSA-N donetidine Chemical compound O1C(CN(C)C)=CC=C1CSCCNC(NC1=O)=NC=C1CC1=CC(=O)NC=C1 MURUHMTVTKOWBY-UHFFFAOYSA-N 0.000 description 1
- 229950004251 dopamantine Drugs 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 229960001857 dopexamine Drugs 0.000 description 1
- RYBJORHCUPVNMB-UHFFFAOYSA-N dopexamine Chemical compound C1=C(O)C(O)=CC=C1CCNCCCCCCNCCC1=CC=CC=C1 RYBJORHCUPVNMB-UHFFFAOYSA-N 0.000 description 1
- 229950006553 dopropidil Drugs 0.000 description 1
- 229950004727 doqualast Drugs 0.000 description 1
- 229950000904 dorastine Drugs 0.000 description 1
- 229950009705 doreptide Drugs 0.000 description 1
- 229950006629 dosergoside Drugs 0.000 description 1
- 229960001393 dosulepin Drugs 0.000 description 1
- LRMJAFKKJLRDLE-UHFFFAOYSA-N dotarizine Chemical compound O1CCOC1(C=1C=CC=CC=1)CCCN(CC1)CCN1C(C=1C=CC=CC=1)C1=CC=CC=C1 LRMJAFKKJLRDLE-UHFFFAOYSA-N 0.000 description 1
- 229950005624 dotarizine Drugs 0.000 description 1
- 229950007375 dotefonium bromide Drugs 0.000 description 1
- 230000002222 downregulating effect Effects 0.000 description 1
- 229960003450 doxacurium chloride Drugs 0.000 description 1
- 229950008058 doxaminol Drugs 0.000 description 1
- 229960002955 doxapram Drugs 0.000 description 1
- 229950004666 doxaprost Drugs 0.000 description 1
- 229960001389 doxazosin Drugs 0.000 description 1
- RUZYUOTYCVRMRZ-UHFFFAOYSA-N doxazosin Chemical compound C1OC2=CC=CC=C2OC1C(=O)N(CC1)CCN1C1=NC(N)=C(C=C(C(OC)=C2)OC)C2=N1 RUZYUOTYCVRMRZ-UHFFFAOYSA-N 0.000 description 1
- 229960003100 doxefazepam Drugs 0.000 description 1
- 229950004453 doxenitoin Drugs 0.000 description 1
- 229960005426 doxepin Drugs 0.000 description 1
- 229950001255 doxibetasol Drugs 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 229960004483 doxofylline Drugs 0.000 description 1
- HWXIGFIVGWUZAO-UHFFFAOYSA-N doxofylline Chemical compound C1=2C(=O)N(C)C(=O)N(C)C=2N=CN1CC1OCCO1 HWXIGFIVGWUZAO-UHFFFAOYSA-N 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 229960002918 doxorubicin hydrochloride Drugs 0.000 description 1
- 229950005497 doxpicomine Drugs 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- HCFDWZZGGLSKEP-UHFFFAOYSA-N doxylamine Chemical compound C=1C=CC=NC=1C(C)(OCCN(C)C)C1=CC=CC=C1 HCFDWZZGGLSKEP-UHFFFAOYSA-N 0.000 description 1
- 229960005178 doxylamine Drugs 0.000 description 1
- UODXSCCNACAPCE-UHFFFAOYSA-N draft:flumetramide Chemical compound C1=CC(C(F)(F)F)=CC=C1C1OCC(=O)NC1 UODXSCCNACAPCE-UHFFFAOYSA-N 0.000 description 1
- 229950005188 dramedilol Drugs 0.000 description 1
- 229950009551 draquinolol Drugs 0.000 description 1
- 229950002266 dribendazole Drugs 0.000 description 1
- 229950009000 drobuline Drugs 0.000 description 1
- GZBONOYGBJSTHF-QLRNAMTQSA-N drocinonide Chemical compound C([C@@H]1CC2)C(=O)CC[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O GZBONOYGBJSTHF-QLRNAMTQSA-N 0.000 description 1
- 229950006082 drocinonide Drugs 0.000 description 1
- 229950001477 droclidinium bromide Drugs 0.000 description 1
- 229950006709 drofenine Drugs 0.000 description 1
- 229950004203 droloxifene Drugs 0.000 description 1
- MCPKSFINULVDNX-UHFFFAOYSA-N drometrizole Chemical compound CC1=CC=C(O)C(N2N=C3C=CC=CC3=N2)=C1 MCPKSFINULVDNX-UHFFFAOYSA-N 0.000 description 1
- 229960000979 drometrizole Drugs 0.000 description 1
- 229940017825 dromostanolone Drugs 0.000 description 1
- 229960004242 dronabinol Drugs 0.000 description 1
- 229950010110 dropempine Drugs 0.000 description 1
- HTAFVGKAHGNWQO-UHFFFAOYSA-N droprenilamine Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)CCNC(C)CC1CCCCC1 HTAFVGKAHGNWQO-UHFFFAOYSA-N 0.000 description 1
- 229950011072 droprenilamine Drugs 0.000 description 1
- 229960004722 dropropizine Drugs 0.000 description 1
- 229950004683 drostanolone propionate Drugs 0.000 description 1
- OMFNSKIUKYOYRG-MOSHPQCFSA-N drotaverine Chemical compound C1=C(OCC)C(OCC)=CC=C1\C=C/1C2=CC(OCC)=C(OCC)C=C2CCN\1 OMFNSKIUKYOYRG-MOSHPQCFSA-N 0.000 description 1
- 229960002065 drotaverine Drugs 0.000 description 1
- 229950005448 drotebanol Drugs 0.000 description 1
- 229950009606 droxacin Drugs 0.000 description 1
- 229950002380 droxicainide Drugs 0.000 description 1
- 229960001850 droxicam Drugs 0.000 description 1
- OEHFRZLKGRKFAS-UHFFFAOYSA-N droxicam Chemical compound C12=CC=CC=C2S(=O)(=O)N(C)C(C2=O)=C1OC(=O)N2C1=CC=CC=N1 OEHFRZLKGRKFAS-UHFFFAOYSA-N 0.000 description 1
- 229960001104 droxidopa Drugs 0.000 description 1
- QXWYKJLNLSIPIN-SFYZADRCSA-N droxidopa Chemical compound OC(=O)[C@H](N)[C@@H](O)C1=CC=C(O)C(O)=C1 QXWYKJLNLSIPIN-SFYZADRCSA-N 0.000 description 1
- FVYYUBRKVVOOAV-UHFFFAOYSA-N droxypropine Chemical compound C=1C=CC=CC=1C1(C(=O)CC)CCN(CCOCCO)CC1 FVYYUBRKVVOOAV-UHFFFAOYSA-N 0.000 description 1
- 229960001949 droxypropine Drugs 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 229950011488 dulofibrate Drugs 0.000 description 1
- 229950005159 dulozafone Drugs 0.000 description 1
- 229950009115 duometacin Drugs 0.000 description 1
- 229950003644 duoperone Drugs 0.000 description 1
- YPUPYVWSTBYCBY-UHFFFAOYSA-N dupracetam Chemical compound C1CCC(=O)N1CC(=O)NNC(=O)CN1CCCC1=O YPUPYVWSTBYCBY-UHFFFAOYSA-N 0.000 description 1
- 229950010707 dupracetam Drugs 0.000 description 1
- 229940105546 durapatite Drugs 0.000 description 1
- BZEWSEKUUPWQDQ-UHFFFAOYSA-N dyclonine Chemical compound C1=CC(OCCCC)=CC=C1C(=O)CCN1CCCCC1 BZEWSEKUUPWQDQ-UHFFFAOYSA-N 0.000 description 1
- 229960000385 dyclonine Drugs 0.000 description 1
- JGMOKGBVKVMRFX-HQZYFCCVSA-N dydrogesterone Chemical compound C1=CC2=CC(=O)CC[C@@]2(C)[C@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 JGMOKGBVKVMRFX-HQZYFCCVSA-N 0.000 description 1
- 229960004913 dydrogesterone Drugs 0.000 description 1
- KSCFJBIXMNOVSH-UHFFFAOYSA-N dyphylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1N(CC(O)CO)C=N2 KSCFJBIXMNOVSH-UHFFFAOYSA-N 0.000 description 1
- 229960001971 ebastine Drugs 0.000 description 1
- MJJALKDDGIKVBE-UHFFFAOYSA-N ebastine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(=O)CCCN1CCC(OC(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 MJJALKDDGIKVBE-UHFFFAOYSA-N 0.000 description 1
- ZQHFZHPUZXNPMF-UHFFFAOYSA-N ebrotidine Chemical compound S1C(N=C(N)N)=NC(CSCCN=CNS(=O)(=O)C=2C=CC(Br)=CC=2)=C1 ZQHFZHPUZXNPMF-UHFFFAOYSA-N 0.000 description 1
- 229950002377 ebrotidine Drugs 0.000 description 1
- 229950010033 ebselen Drugs 0.000 description 1
- 229950005111 ecastolol Drugs 0.000 description 1
- 229960002445 echothiophate iodide Drugs 0.000 description 1
- 229950008981 ecipramidil Drugs 0.000 description 1
- 229950000627 eclanamine Drugs 0.000 description 1
- YCRFSKUCDBJWLX-HUUCEWRRSA-N eclanamine Chemical compound C=1C=C(Cl)C(Cl)=CC=1N(C(=O)CC)[C@@H]1CCC[C@H]1N(C)C YCRFSKUCDBJWLX-HUUCEWRRSA-N 0.000 description 1
- 229950001263 eclazolast Drugs 0.000 description 1
- 229960003913 econazole Drugs 0.000 description 1
- OVXQHPWHMXOFRD-UHFFFAOYSA-M ecothiopate iodide Chemical compound [I-].CCOP(=O)(OCC)SCC[N+](C)(C)C OVXQHPWHMXOFRD-UHFFFAOYSA-M 0.000 description 1
- 229950008272 ectylurea Drugs 0.000 description 1
- 229950011461 edelfosine Drugs 0.000 description 1
- 229960001484 edetic acid Drugs 0.000 description 1
- 229960000226 edetol Drugs 0.000 description 1
- HWXXPLRSBHPGSF-KNOXWWKRSA-N edifolone Chemical compound C([C@@H]1[C@@H]([C@]2(CC3)CCN)CC[C@]4([C@H]1CCC41OCCO1)C)C=C2CC13OCCO1 HWXXPLRSBHPGSF-KNOXWWKRSA-N 0.000 description 1
- 229950007218 edifolone Drugs 0.000 description 1
- UOYMGHSCINLOBK-ZKKYLISVSA-N edogestrone Chemical compound C([C@H]1[C@@H]2CC[C@]([C@]2(CC[C@@H]1[C@@]1(C)CC2)C)(OC(=O)C)C(C)=O)C(C)=C1CC12OCCO1 UOYMGHSCINLOBK-ZKKYLISVSA-N 0.000 description 1
- 229950010837 edogestrone Drugs 0.000 description 1
- XACKNLSZYYIACO-DJLDLDEBSA-N edoxudine Chemical compound O=C1NC(=O)C(CC)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XACKNLSZYYIACO-DJLDLDEBSA-N 0.000 description 1
- 229960002030 edoxudine Drugs 0.000 description 1
- 229950001765 efaroxan Drugs 0.000 description 1
- 229950001454 efetozole Drugs 0.000 description 1
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical compound NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 description 1
- 229960002759 eflornithine Drugs 0.000 description 1
- 229960003859 efloxate Drugs 0.000 description 1
- 229950003445 efrotomycin Drugs 0.000 description 1
- 235000013345 egg yolk Nutrition 0.000 description 1
- 210000002969 egg yolk Anatomy 0.000 description 1
- 229950010281 elantrine Drugs 0.000 description 1
- 229950008990 elanzepine Drugs 0.000 description 1
- 238000007336 electrophilic substitution reaction Methods 0.000 description 1
- BSPSXMXQKZZNFP-UHFFFAOYSA-N elfazepam Chemical compound N=1CC(=O)N(CCS(=O)(=O)CC)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F BSPSXMXQKZZNFP-UHFFFAOYSA-N 0.000 description 1
- 229950008850 elfazepam Drugs 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229960002852 ellagic acid Drugs 0.000 description 1
- 235000004132 ellagic acid Nutrition 0.000 description 1
- 229950000549 elliptinium acetate Drugs 0.000 description 1
- 229950003860 elmustine Drugs 0.000 description 1
- 229950010010 elnadipine Drugs 0.000 description 1
- 229950009219 eltenac Drugs 0.000 description 1
- 229950006047 eltoprazine Drugs 0.000 description 1
- WVLHGCRWEHCIOT-UHFFFAOYSA-N eltoprazine Chemical compound C1CNCCN1C1=CC=CC2=C1OCCO2 WVLHGCRWEHCIOT-UHFFFAOYSA-N 0.000 description 1
- 229950002472 elucaine Drugs 0.000 description 1
- 229950010923 elziverine Drugs 0.000 description 1
- 229950000472 embramine Drugs 0.000 description 1
- URSRSKSNFPUKGH-UHFFFAOYSA-N embramine Chemical compound C=1C=C(Br)C=CC=1C(C)(OCCN(C)C)C1=CC=CC=C1 URSRSKSNFPUKGH-UHFFFAOYSA-N 0.000 description 1
- 229950009082 embutramide Drugs 0.000 description 1
- LMBMDLOSPKIWAP-UHFFFAOYSA-N embutramide Chemical compound OCCCC(=O)NCC(CC)(CC)C1=CC=CC(OC)=C1 LMBMDLOSPKIWAP-UHFFFAOYSA-N 0.000 description 1
- 229960002493 emepronium bromide Drugs 0.000 description 1
- 229960002694 emetine Drugs 0.000 description 1
- AUVVAXYIELKVAI-CKBKHPSWSA-N emetine Chemical compound N1CCC2=CC(OC)=C(OC)C=C2[C@H]1C[C@H]1C[C@H]2C3=CC(OC)=C(OC)C=C3CCN2C[C@@H]1CC AUVVAXYIELKVAI-CKBKHPSWSA-N 0.000 description 1
- AUVVAXYIELKVAI-UWBTVBNJSA-N emetine Natural products N1CCC2=CC(OC)=C(OC)C=C2[C@H]1C[C@H]1C[C@H]2C3=CC(OC)=C(OC)C=C3CCN2C[C@H]1CC AUVVAXYIELKVAI-UWBTVBNJSA-N 0.000 description 1
- 229950000269 emiglitate Drugs 0.000 description 1
- 229950010243 emorfazone Drugs 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- SLWGJZPKHAXZQL-UHFFFAOYSA-N emylcamate Chemical compound CCC(C)(CC)OC(N)=O SLWGJZPKHAXZQL-UHFFFAOYSA-N 0.000 description 1
- 229960000650 emylcamate Drugs 0.000 description 1
- 229960002680 enalaprilat Drugs 0.000 description 1
- LZFZMUMEGBBDTC-QEJZJMRPSA-N enalaprilat (anhydrous) Chemical compound C([C@H](N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 LZFZMUMEGBBDTC-QEJZJMRPSA-N 0.000 description 1
- 229950010048 enbucrilate Drugs 0.000 description 1
- JJJFUHOGVZWXNQ-UHFFFAOYSA-N enbucrilate Chemical compound CCCCOC(=O)C(=C)C#N JJJFUHOGVZWXNQ-UHFFFAOYSA-N 0.000 description 1
- PJWPNDMDCLXCOM-UHFFFAOYSA-N encainide Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=CC=C1CCC1N(C)CCCC1 PJWPNDMDCLXCOM-UHFFFAOYSA-N 0.000 description 1
- 229960001142 encainide Drugs 0.000 description 1
- 229950010052 enciprazine Drugs 0.000 description 1
- 229950002791 encyprate Drugs 0.000 description 1
- 229950007989 endomide Drugs 0.000 description 1
- 210000003989 endothelium vascular Anatomy 0.000 description 1
- ALAXZYHFVBSJKZ-UHFFFAOYSA-N endralazine Chemical compound C1CC=2N=NC(NN)=CC=2CN1C(=O)C1=CC=CC=C1 ALAXZYHFVBSJKZ-UHFFFAOYSA-N 0.000 description 1
- 229960002029 endralazine Drugs 0.000 description 1
- 229950004233 enefexine Drugs 0.000 description 1
- 229950005641 enestebol Drugs 0.000 description 1
- 229950010996 enfenamic acid Drugs 0.000 description 1
- 229960000305 enflurane Drugs 0.000 description 1
- JPGQOUSTVILISH-UHFFFAOYSA-N enflurane Chemical compound FC(F)OC(F)(F)C(F)Cl JPGQOUSTVILISH-UHFFFAOYSA-N 0.000 description 1
- 229950004266 eniclobrate Drugs 0.000 description 1
- 229960002125 enilconazole Drugs 0.000 description 1
- 229950002012 enilospirone Drugs 0.000 description 1
- 229950007459 enisoprost Drugs 0.000 description 1
- 229950011487 enocitabine Drugs 0.000 description 1
- 229950002107 enolicam Drugs 0.000 description 1
- 229960002549 enoxacin Drugs 0.000 description 1
- IDYZIJYBMGIQMJ-UHFFFAOYSA-N enoxacin Chemical compound N1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 IDYZIJYBMGIQMJ-UHFFFAOYSA-N 0.000 description 1
- 229950001183 enoxamast Drugs 0.000 description 1
- 229960000972 enoximone Drugs 0.000 description 1
- ZJKNESGOIKRXQY-UHFFFAOYSA-N enoximone Chemical compound C1=CC(SC)=CC=C1C(=O)C1=C(C)NC(=O)N1 ZJKNESGOIKRXQY-UHFFFAOYSA-N 0.000 description 1
- 229960003720 enoxolone Drugs 0.000 description 1
- VXCSCOXOGWUYKK-UHFFFAOYSA-N enprazepine Chemical compound CN(C)CCCN1CC2=CC=CC=C2C(=C)C2=CC=CC=C12 VXCSCOXOGWUYKK-UHFFFAOYSA-N 0.000 description 1
- 229950009212 enprazepine Drugs 0.000 description 1
- QVDKSPUZWYTNQA-UHFFFAOYSA-N enprofylline Chemical compound O=C1NC(=O)N(CCC)C2=NC=N[C]21 QVDKSPUZWYTNQA-UHFFFAOYSA-N 0.000 description 1
- 229950000579 enprofylline Drugs 0.000 description 1
- 229950001022 enpromate Drugs 0.000 description 1
- 229960003559 enprostil Drugs 0.000 description 1
- 229960000740 enrofloxacin Drugs 0.000 description 1
- 229960001986 entsufon sodium Drugs 0.000 description 1
- 229950000219 enviomycin Drugs 0.000 description 1
- 229950000529 enviradene Drugs 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229960002711 epanolol Drugs 0.000 description 1
- 229960002565 eperisone Drugs 0.000 description 1
- 229960002179 ephedrine Drugs 0.000 description 1
- 229950002864 epicainide Drugs 0.000 description 1
- 229960002457 epicillin Drugs 0.000 description 1
- RPBAFSBGYDKNRG-NJBDSQKTSA-N epicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CCC=CC1 RPBAFSBGYDKNRG-NJBDSQKTSA-N 0.000 description 1
- SBFXHXZNBNFPHV-PXXBSISHSA-N epicriptine Chemical compound C1=CC([C@H]2C[C@H](CN(C)[C@@H]2C2)C(=O)N[C@]3(C(=O)N4[C@H](C(N5CCC[C@H]5[C@]4(O)O3)=O)[C@H](C)CC)C(C)C)=C3C2=CNC3=C1 SBFXHXZNBNFPHV-PXXBSISHSA-N 0.000 description 1
- 229950007315 epiestriol Drugs 0.000 description 1
- 229960004595 epimestrol Drugs 0.000 description 1
- 229960003449 epinastine Drugs 0.000 description 1
- WHWZLSFABNNENI-UHFFFAOYSA-N epinastine Chemical compound C1C2=CC=CC=C2C2CN=C(N)N2C2=CC=CC=C21 WHWZLSFABNNENI-UHFFFAOYSA-N 0.000 description 1
- 229960005139 epinephrine Drugs 0.000 description 1
- 229960000404 epinephryl borate Drugs 0.000 description 1
- 229950004926 epipropidine Drugs 0.000 description 1
- 229950003801 epirizole Drugs 0.000 description 1
- 229950011561 epiroprim Drugs 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229950002973 epitiostanol Drugs 0.000 description 1
- RINBGYCKMGDWPY-UHFFFAOYSA-N epitizide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NC(CSCC(F)(F)F)NS2(=O)=O RINBGYCKMGDWPY-UHFFFAOYSA-N 0.000 description 1
- 229950010350 epitizide Drugs 0.000 description 1
- 229960001123 epoprostenol Drugs 0.000 description 1
- KAQKFAOMNZTLHT-VVUHWYTRSA-N epoprostenol Chemical compound O1C(=CCCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@H]21 KAQKFAOMNZTLHT-VVUHWYTRSA-N 0.000 description 1
- 229950002674 epostane Drugs 0.000 description 1
- CETKWEWBSMKADK-GSXVSZIWSA-N epostane Chemical compound C([C@]1(C)[C@@](C)(O)CC[C@H]1[C@@H]1CC2)C[C@@H]1[C@]1(C)[C@]32O[C@]3(C)C(O)=C(C#N)C1 CETKWEWBSMKADK-GSXVSZIWSA-N 0.000 description 1
- 229960002561 eprazinone Drugs 0.000 description 1
- BSHWLCACYCVCJE-UHFFFAOYSA-N eprazinone Chemical compound C=1C=CC=CC=1C(OCC)CN(CC1)CCN1CC(C)C(=O)C1=CC=CC=C1 BSHWLCACYCVCJE-UHFFFAOYSA-N 0.000 description 1
- 229950005711 eprovafen Drugs 0.000 description 1
- 229950011080 eproxindine Drugs 0.000 description 1
- 229960002338 eprozinol Drugs 0.000 description 1
- QSRHLIUOSXVKTG-UHFFFAOYSA-N eprozinol Chemical compound C=1C=CC=CC=1C(OC)CN(CC1)CCN1CCC(O)C1=CC=CC=C1 QSRHLIUOSXVKTG-UHFFFAOYSA-N 0.000 description 1
- LGUDKOQUWIHXOV-UHFFFAOYSA-N epsiprantel Chemical compound C1C(C2=CC=CC=C2CCC2)N2C(=O)CN1C(=O)C1CCCCC1 LGUDKOQUWIHXOV-UHFFFAOYSA-N 0.000 description 1
- 229960005362 epsiprantel Drugs 0.000 description 1
- 229950006293 eptaloprost Drugs 0.000 description 1
- ZOWQTJXNFTWSCS-IAQYHMDHSA-N eptazocine Chemical compound C1N(C)CC[C@@]2(C)C3=CC(O)=CC=C3C[C@@H]1C2 ZOWQTJXNFTWSCS-IAQYHMDHSA-N 0.000 description 1
- 229950010920 eptazocine Drugs 0.000 description 1
- WKRLQDKEXYKHJB-HFTRVMKXSA-N equilin Chemical compound OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4C3=CCC2=C1 WKRLQDKEXYKHJB-HFTRVMKXSA-N 0.000 description 1
- 229960003262 erdosteine Drugs 0.000 description 1
- QGFORSXNKQLDNO-UHFFFAOYSA-N erdosteine Chemical compound OC(=O)CSCC(=O)NC1CCSC1=O QGFORSXNKQLDNO-UHFFFAOYSA-N 0.000 description 1
- 229960002061 ergocalciferol Drugs 0.000 description 1
- 229940040520 ergoloid mesylates Drugs 0.000 description 1
- 229960001405 ergometrine Drugs 0.000 description 1
- DNVPQKQSNYMLRS-SOWFXMKYSA-N ergosterol Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H](CC[C@]3([C@H]([C@H](C)/C=C/[C@@H](C)C(C)C)CC[C@H]33)C)C3=CC=C21 DNVPQKQSNYMLRS-SOWFXMKYSA-N 0.000 description 1
- 229960004943 ergotamine Drugs 0.000 description 1
- OFKDAAIKGIBASY-VFGNJEKYSA-N ergotamine Chemical compound C([C@H]1C(=O)N2CCC[C@H]2[C@]2(O)O[C@@](C(N21)=O)(C)NC(=O)[C@H]1CN([C@H]2C(C3=CC=CC4=NC=C([C]34)C2)=C1)C)C1=CC=CC=C1 OFKDAAIKGIBASY-VFGNJEKYSA-N 0.000 description 1
- XCGSFFUVFURLIX-UHFFFAOYSA-N ergotaminine Natural products C1=C(C=2C=CC=C3NC=C(C=23)C2)C2N(C)CC1C(=O)NC(C(N12)=O)(C)OC1(O)C1CCCN1C(=O)C2CC1=CC=CC=C1 XCGSFFUVFURLIX-UHFFFAOYSA-N 0.000 description 1
- 229950007410 ericolol Drugs 0.000 description 1
- 229960005450 eritrityl tetranitrate Drugs 0.000 description 1
- 229950007947 erizepine Drugs 0.000 description 1
- SGMXCAKSGHLWBE-UHFFFAOYSA-N erizepine Chemical compound C12=CC=CC=C2N(C)C2=CC=CC=C2C2=C1CCN(C)CC2 SGMXCAKSGHLWBE-UHFFFAOYSA-N 0.000 description 1
- 229950000108 erocainide Drugs 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- TYQXKHPOXXXCTP-CSLYCKPJSA-N erythromycin A 2'-propanoate Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)OC(=O)CC)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 TYQXKHPOXXXCTP-CSLYCKPJSA-N 0.000 description 1
- IDRYSCOQVVUBIJ-PPGFLMPOSA-N erythromycin B Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@H]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)C)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 IDRYSCOQVVUBIJ-PPGFLMPOSA-N 0.000 description 1
- 229950007610 erythromycin acistrate Drugs 0.000 description 1
- 229960000741 erythromycin ethylsuccinate Drugs 0.000 description 1
- NSYZCCDSJNWWJL-YXOIYICCSA-N erythromycin ethylsuccinate Chemical compound O1[C@H](C)C[C@H](N(C)C)[C@@H](OC(=O)CCC(=O)OCC)[C@@H]1O[C@H]1[C@@](O)(C)C[C@@H](C)C(=O)[C@H](C)[C@@H](O)[C@](C)(O)[C@@H](CC)OC(=O)[C@H](C)[C@@H](O[C@@H]2O[C@@H](C)[C@H](O)[C@](C)(OC)C2)[C@@H]1C NSYZCCDSJNWWJL-YXOIYICCSA-N 0.000 description 1
- 229950001028 erythromycin propionate Drugs 0.000 description 1
- IINNWAYUJNWZRM-UHFFFAOYSA-L erythrosin B Chemical compound [Na+].[Na+].[O-]C(=O)C1=CC=CC=C1C1=C2C=C(I)C(=O)C(I)=C2OC2=C(I)C([O-])=C(I)C=C21 IINNWAYUJNWZRM-UHFFFAOYSA-L 0.000 description 1
- 229940011411 erythrosine Drugs 0.000 description 1
- 239000004174 erythrosine Substances 0.000 description 1
- 235000012732 erythrosine Nutrition 0.000 description 1
- 229950006349 esaprazole Drugs 0.000 description 1
- 229950010051 esculamine Drugs 0.000 description 1
- 229950003340 eseridine Drugs 0.000 description 1
- 229950006887 esflurbiprofen Drugs 0.000 description 1
- AQNDDEOPVVGCPG-UHFFFAOYSA-N esmolol Chemical compound COC(=O)CCC1=CC=C(OCC(O)CNC(C)C)C=C1 AQNDDEOPVVGCPG-UHFFFAOYSA-N 0.000 description 1
- 229960003745 esmolol Drugs 0.000 description 1
- ITSGNOIFAJAQHJ-BMFNZSJVSA-N esorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)C[C@H](C)O1 ITSGNOIFAJAQHJ-BMFNZSJVSA-N 0.000 description 1
- 229950002017 esorubicin Drugs 0.000 description 1
- CDCHDCWJMGXXRH-UHFFFAOYSA-N estazolam Chemical compound C=1C(Cl)=CC=C(N2C=NN=C2CN=2)C=1C=2C1=CC=CC=C1 CDCHDCWJMGXXRH-UHFFFAOYSA-N 0.000 description 1
- 229960002336 estazolam Drugs 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 229950002007 estradiol benzoate Drugs 0.000 description 1
- 229960005416 estradiol cypionate Drugs 0.000 description 1
- 229950010215 estradiol dipropionate Drugs 0.000 description 1
- 229950004659 estradiol enanthate Drugs 0.000 description 1
- RFWTZQAOOLFXAY-BZDYCCQFSA-N estradiol enanthate Chemical compound C1CC2=CC(O)=CC=C2[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CCCCCC)[C@@]1(C)CC2 RFWTZQAOOLFXAY-BZDYCCQFSA-N 0.000 description 1
- 229950005281 estradiol undecylate Drugs 0.000 description 1
- TXHUMRBWIWWBGW-GVGNIZHQSA-N estradiol undecylate Chemical compound C1CC2=CC(O)=CC=C2[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CCCCCCCCCC)[C@@]1(C)CC2 TXHUMRBWIWWBGW-GVGNIZHQSA-N 0.000 description 1
- 229960004766 estradiol valerate Drugs 0.000 description 1
- 229960004750 estramustine phosphate Drugs 0.000 description 1
- ADFOJJHRTBFFOF-RBRWEJTLSA-N estramustine phosphate Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)OP(O)(O)=O)[C@@H]4[C@@H]3CCC2=C1 ADFOJJHRTBFFOF-RBRWEJTLSA-N 0.000 description 1
- 229950008961 estrapronicate Drugs 0.000 description 1
- NTHOJXSFNBUDMY-ZRNYENFQSA-N estrazinol Chemical compound C1C[C@]2(C)[C@@](C#C)(O)CC[C@H]2N2CCC3=CC(OC)=CC=C3[C@H]21 NTHOJXSFNBUDMY-ZRNYENFQSA-N 0.000 description 1
- 229950010261 estrazinol Drugs 0.000 description 1
- PROQIPRRNZUXQM-ZXXIGWHRSA-N estriol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H]([C@H](O)C4)O)[C@@H]4[C@@H]3CCC2=C1 PROQIPRRNZUXQM-ZXXIGWHRSA-N 0.000 description 1
- 229960001348 estriol Drugs 0.000 description 1
- 229950010129 estrofurate Drugs 0.000 description 1
- 229960003399 estrone Drugs 0.000 description 1
- JKKFKPJIXZFSSB-CBZIJGRNSA-N estrone 3-sulfate Chemical compound OS(=O)(=O)OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 JKKFKPJIXZFSSB-CBZIJGRNSA-N 0.000 description 1
- 229940081345 estropipate Drugs 0.000 description 1
- HZEQBCVBILBTEP-ZFINNJDLSA-N estropipate Chemical compound C1CNCCN1.OS(=O)(=O)OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 HZEQBCVBILBTEP-ZFINNJDLSA-N 0.000 description 1
- CHDGAVDQRSPBTA-UHFFFAOYSA-N esuprone Chemical compound CC1=C(C)C(=O)OC2=CC(OS(=O)(=O)CC)=CC=C21 CHDGAVDQRSPBTA-UHFFFAOYSA-N 0.000 description 1
- 229950007673 esuprone Drugs 0.000 description 1
- 229950008786 etabenzarone Drugs 0.000 description 1
- 229950010835 etacepride Drugs 0.000 description 1
- 229960003199 etacrynic acid Drugs 0.000 description 1
- AVOLMBLBETYQHX-UHFFFAOYSA-N etacrynic acid Chemical compound CCC(=C)C(=O)C1=CC=C(OCC(O)=O)C(Cl)=C1Cl AVOLMBLBETYQHX-UHFFFAOYSA-N 0.000 description 1
- IRVLBORJKFZWMI-JQWIXIFHSA-N etafedrine Chemical compound CCN(C)[C@@H](C)[C@H](O)C1=CC=CC=C1 IRVLBORJKFZWMI-JQWIXIFHSA-N 0.000 description 1
- 229950002456 etafedrine Drugs 0.000 description 1
- OEGDFSLNGABBKJ-UHFFFAOYSA-N etafenone Chemical compound CCN(CC)CCOC1=CC=CC=C1C(=O)CCC1=CC=CC=C1 OEGDFSLNGABBKJ-UHFFFAOYSA-N 0.000 description 1
- 229960004351 etafenone Drugs 0.000 description 1
- AWKLBIOQCIORSB-UHFFFAOYSA-N etamiphylline Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2CCN(CC)CC AWKLBIOQCIORSB-UHFFFAOYSA-N 0.000 description 1
- 229960000505 etamiphylline Drugs 0.000 description 1
- 229960005180 etamivan Drugs 0.000 description 1
- BQJODPIMMWWMFC-UHFFFAOYSA-N etamivan Chemical compound CCN(CC)C(=O)C1=CC=C(O)C(OC)=C1 BQJODPIMMWWMFC-UHFFFAOYSA-N 0.000 description 1
- 229950004798 etamocycline Drugs 0.000 description 1
- 229950006566 etanidazole Drugs 0.000 description 1
- WCDWBPCFGJXFJZ-UHFFFAOYSA-N etanidazole Chemical compound OCCNC(=O)CN1C=CN=C1[N+]([O-])=O WCDWBPCFGJXFJZ-UHFFFAOYSA-N 0.000 description 1
- 229950002751 etanterol Drugs 0.000 description 1
- UVTJKLLUVOTSOB-UHFFFAOYSA-N etaqualone Chemical compound CCC1=CC=CC=C1N1C(=O)C2=CC=CC=C2N=C1C UVTJKLLUVOTSOB-UHFFFAOYSA-N 0.000 description 1
- 229950010472 etaqualone Drugs 0.000 description 1
- 229950001682 etasuline Drugs 0.000 description 1
- BLGFGFHRMMDRPC-UHFFFAOYSA-N etazepine Chemical compound CN1C(=O)C2=CC=CC=C2C(OCC)C2=CC=CC=C21 BLGFGFHRMMDRPC-UHFFFAOYSA-N 0.000 description 1
- 229950000201 etazepine Drugs 0.000 description 1
- 229950009329 etazolate Drugs 0.000 description 1
- OPQRBXUBWHDHPQ-UHFFFAOYSA-N etazolate Chemical compound CCOC(=O)C1=CN=C2N(CC)N=CC2=C1NN=C(C)C OPQRBXUBWHDHPQ-UHFFFAOYSA-N 0.000 description 1
- 229950008342 etebenecid Drugs 0.000 description 1
- UACOQEQOBAQRDQ-UHFFFAOYSA-N etebenecid Chemical compound CCN(CC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 UACOQEQOBAQRDQ-UHFFFAOYSA-N 0.000 description 1
- DACOQFZGGLCXMA-UHFFFAOYSA-N eterobarb Chemical compound C=1C=CC=CC=1C1(CC)C(=O)N(COC)C(=O)N(COC)C1=O DACOQFZGGLCXMA-UHFFFAOYSA-N 0.000 description 1
- 229950009327 eterobarb Drugs 0.000 description 1
- 229950006159 etersalate Drugs 0.000 description 1
- PXBFSRVXEKCBFP-UHFFFAOYSA-N etersalate Chemical compound C1=CC(NC(=O)C)=CC=C1OCCOC(=O)C1=CC=CC=C1OC(C)=O PXBFSRVXEKCBFP-UHFFFAOYSA-N 0.000 description 1
- 229960001588 ethacridine Drugs 0.000 description 1
- CIKWKGFPFXJVGW-UHFFFAOYSA-N ethacridine Chemical compound C1=C(N)C=CC2=C(N)C3=CC(OCC)=CC=C3N=C21 CIKWKGFPFXJVGW-UHFFFAOYSA-N 0.000 description 1
- 229940011899 ethamsylate Drugs 0.000 description 1
- 229940031124 ethanolamine oleate Drugs 0.000 description 1
- HYSIJEPDMLSIQJ-UHFFFAOYSA-N ethanolate;1-phenylbutane-1,3-dione;titanium(4+) Chemical compound [Ti+4].CC[O-].CC[O-].CC(=O)[CH-]C(=O)C1=CC=CC=C1.CC(=O)[CH-]C(=O)C1=CC=CC=C1 HYSIJEPDMLSIQJ-UHFFFAOYSA-N 0.000 description 1
- 229960005269 ethaverine Drugs 0.000 description 1
- 229960000514 ethenzamide Drugs 0.000 description 1
- SBNKFTQSBPKMBZ-UHFFFAOYSA-N ethenzamide Chemical compound CCOC1=CC=CC=C1C(N)=O SBNKFTQSBPKMBZ-UHFFFAOYSA-N 0.000 description 1
- 229960002568 ethinylestradiol Drugs 0.000 description 1
- WGJHHMKQBWSQIY-UHFFFAOYSA-N ethoheptazine Chemical compound C=1C=CC=CC=1C1(C(=O)OCC)CCCN(C)CC1 WGJHHMKQBWSQIY-UHFFFAOYSA-N 0.000 description 1
- 229960000569 ethoheptazine Drugs 0.000 description 1
- 229950000322 ethomoxane Drugs 0.000 description 1
- 229960002767 ethosuximide Drugs 0.000 description 1
- HAPOVYFOVVWLRS-UHFFFAOYSA-N ethosuximide Chemical compound CCC1(C)CC(=O)NC1=O HAPOVYFOVVWLRS-UHFFFAOYSA-N 0.000 description 1
- SZQIFWWUIBRPBZ-UHFFFAOYSA-N ethotoin Chemical compound O=C1N(CC)C(=O)NC1C1=CC=CC=C1 SZQIFWWUIBRPBZ-UHFFFAOYSA-N 0.000 description 1
- 229960003533 ethotoin Drugs 0.000 description 1
- 229950001342 ethoxazorutoside Drugs 0.000 description 1
- OUZWUKMCLIBBOG-UHFFFAOYSA-N ethoxzolamide Chemical compound CCOC1=CC=C2N=C(S(N)(=O)=O)SC2=C1 OUZWUKMCLIBBOG-UHFFFAOYSA-N 0.000 description 1
- 229950005098 ethoxzolamide Drugs 0.000 description 1
- GBPZYMBDOBODNK-SFTDATJTSA-N ethyl (2s)-2-[[(2s)-2-acetamido-3-[4-[bis(2-chloroethyl)amino]phenyl]propanoyl]amino]-4-methylpentanoate Chemical compound CCOC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(C)=O)CC1=CC=C(N(CCCl)CCCl)C=C1 GBPZYMBDOBODNK-SFTDATJTSA-N 0.000 description 1
- ZCKKHYXUQFTBIK-XPSMFNQNSA-N ethyl (2z)-2-[(5s)-3-methyl-4-oxo-5-piperidin-1-yl-1,3-thiazolidin-2-ylidene]acetate Chemical compound O=C1N(C)C(=C/C(=O)OCC)/S[C@@H]1N1CCCCC1 ZCKKHYXUQFTBIK-XPSMFNQNSA-N 0.000 description 1
- IAJNXBNRYMEYAZ-UHFFFAOYSA-N ethyl 2-cyano-3,3-diphenylprop-2-enoate Chemical group C=1C=CC=CC=1C(=C(C#N)C(=O)OCC)C1=CC=CC=C1 IAJNXBNRYMEYAZ-UHFFFAOYSA-N 0.000 description 1
- NWWORXYTJRPSMC-QKPAOTATSA-N ethyl 4-[2-[(2r,3r,4r,5s)-3,4,5-trihydroxy-2-(hydroxymethyl)piperidin-1-yl]ethoxy]benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1OCCN1[C@H](CO)[C@@H](O)[C@H](O)[C@@H](O)C1 NWWORXYTJRPSMC-QKPAOTATSA-N 0.000 description 1
- LVVXOXRUTDAKFE-UHFFFAOYSA-N ethyl 6,7-bis(2-methylpropoxy)-4-oxo-1h-quinoline-3-carboxylate Chemical compound CC(C)COC1=C(OCC(C)C)C=C2C(=O)C(C(=O)OCC)=CNC2=C1 LVVXOXRUTDAKFE-UHFFFAOYSA-N 0.000 description 1
- JBCLGHRMQCEPFM-UHFFFAOYSA-N ethyl 6,7-bis(cyclopropylmethoxy)-4-oxo-1h-quinoline-3-carboxylate Chemical compound C1CC1COC=1C=C2C(=O)C(C(=O)OCC)=CNC2=CC=1OCC1CC1 JBCLGHRMQCEPFM-UHFFFAOYSA-N 0.000 description 1
- 229960002822 ethyl biscoumacetate Drugs 0.000 description 1
- SEGSDVUVOWIWFX-UHFFFAOYSA-N ethyl biscoumacetate Chemical compound C1=CC=C2C(=O)C(C(C=3C(C4=CC=CC=C4OC=3O)=O)C(=O)OCC)=C(O)OC2=C1 SEGSDVUVOWIWFX-UHFFFAOYSA-N 0.000 description 1
- PUJLIQLPZOZCOP-UHFFFAOYSA-N ethyl carfluzepate Chemical compound C12=CC(Cl)=CC=C2N(C(=O)NC)C(=O)C(C(=O)OCC)N=C1C1=CC=CC=C1F PUJLIQLPZOZCOP-UHFFFAOYSA-N 0.000 description 1
- 229950004336 ethyl carfluzepate Drugs 0.000 description 1
- 229950010890 ethyl cartrizoate Drugs 0.000 description 1
- ZAMACTJOCIFTPJ-UHFFFAOYSA-N ethyl dibunate Chemical compound CC(C)(C)C1=CC=C2C(S(=O)(=O)OCC)=CC(C(C)(C)C)=CC2=C1 ZAMACTJOCIFTPJ-UHFFFAOYSA-N 0.000 description 1
- 229950001503 ethyl dibunate Drugs 0.000 description 1
- XFVPZJMXRNBHLQ-UHFFFAOYSA-N ethyl dirazepate Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(C(=O)OCC)N=C1C1=CC=CC=C1Cl XFVPZJMXRNBHLQ-UHFFFAOYSA-N 0.000 description 1
- 229950000935 ethyl dirazepate Drugs 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 229960001617 ethyl hydroxybenzoate Drugs 0.000 description 1
- 229960004759 ethyl loflazepate Drugs 0.000 description 1
- NBODAQXRWMHEBP-UHFFFAOYSA-N ethyl n-[2-(dimethylamino)ethyl]-n-[3-(trifluoromethyl)phenyl]carbamate Chemical compound CCOC(=O)N(CCN(C)C)C1=CC=CC(C(F)(F)F)=C1 NBODAQXRWMHEBP-UHFFFAOYSA-N 0.000 description 1
- OGXBVBBMMWSZJO-UHFFFAOYSA-N ethyl n-benzyl-n-cyclopropylcarbamate Chemical compound C1CC1N(C(=O)OCC)CC1=CC=CC=C1 OGXBVBBMMWSZJO-UHFFFAOYSA-N 0.000 description 1
- 235000010228 ethyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004403 ethyl p-hydroxybenzoate Substances 0.000 description 1
- IOCHDYUJEBGIKB-UHFFFAOYSA-M ethyl-[(3-methoxyphenyl)methyl]-dimethylazanium;4-methylbenzenesulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1.CC[N+](C)(C)CC1=CC=CC(OC)=C1 IOCHDYUJEBGIKB-UHFFFAOYSA-M 0.000 description 1
- UPCIBFUJJLCOQG-UHFFFAOYSA-L ethyl-[2-[2-[ethyl(dimethyl)azaniumyl]ethyl-methylamino]ethyl]-dimethylazanium;dibromide Chemical compound [Br-].[Br-].CC[N+](C)(C)CCN(C)CC[N+](C)(C)CC UPCIBFUJJLCOQG-UHFFFAOYSA-L 0.000 description 1
- QKODANAAHZUYET-UHFFFAOYSA-N ethylantimony Chemical compound CC[Sb] QKODANAAHZUYET-UHFFFAOYSA-N 0.000 description 1
- IYBKWXQWKPSYDT-UHFFFAOYSA-L ethylene glycol disuccinate bis(sulfo-N-succinimidyl) ester sodium salt Chemical compound [Na+].[Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)CCC(=O)OCCOC(=O)CCC(=O)ON1C(=O)C(S([O-])(=O)=O)CC1=O IYBKWXQWKPSYDT-UHFFFAOYSA-L 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 229960001460 ethylestrenol Drugs 0.000 description 1
- AOXRBFRFYPMWLR-XGXHKTLJSA-N ethylestrenol Chemical compound C1CC2=CCCC[C@@H]2[C@@H]2[C@@H]1[C@@H]1CC[C@](CC)(O)[C@@]1(C)CC2 AOXRBFRFYPMWLR-XGXHKTLJSA-N 0.000 description 1
- 229950003871 ethylhydrocupreine Drugs 0.000 description 1
- MORSAEFGQPDBKM-UHFFFAOYSA-N ethylmethylthiambutene Chemical compound C=1C=CSC=1C(=CC(C)N(C)CC)C1=CC=CS1 MORSAEFGQPDBKM-UHFFFAOYSA-N 0.000 description 1
- 229950006111 ethylmethylthiambutene Drugs 0.000 description 1
- 229960004578 ethylmorphine Drugs 0.000 description 1
- LENNRXOJHWNHSD-UHFFFAOYSA-N ethylnorepinephrine Chemical compound CCC(N)C(O)C1=CC=C(O)C(O)=C1 LENNRXOJHWNHSD-UHFFFAOYSA-N 0.000 description 1
- 229960002267 ethylnorepinephrine Drugs 0.000 description 1
- NUVBSKCKDOMJSU-UHFFFAOYSA-N ethylparaben Chemical compound CCOC(=O)C1=CC=C(O)C=C1 NUVBSKCKDOMJSU-UHFFFAOYSA-N 0.000 description 1
- 229950007424 ethynerone Drugs 0.000 description 1
- ONKUMRGIYFNPJW-KIEAKMPYSA-N ethynodiol diacetate Chemical compound C1C[C@]2(C)[C@@](C#C)(OC(C)=O)CC[C@H]2[C@@H]2CCC3=C[C@@H](OC(=O)C)CC[C@@H]3[C@H]21 ONKUMRGIYFNPJW-KIEAKMPYSA-N 0.000 description 1
- 229940012028 ethynodiol diacetate Drugs 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229950010952 ethypicone Drugs 0.000 description 1
- 229950003103 etibendazole Drugs 0.000 description 1
- 229950007535 eticlopride Drugs 0.000 description 1
- IFYLVUHLOOCYBG-UHFFFAOYSA-N eticyclidine Chemical compound C=1C=CC=CC=1C1(NCC)CCCCC1 IFYLVUHLOOCYBG-UHFFFAOYSA-N 0.000 description 1
- 229950005343 eticyclidine Drugs 0.000 description 1
- 229960004585 etidronic acid Drugs 0.000 description 1
- WNKCJOWTKXGERE-UHFFFAOYSA-N etifelmine Chemical compound C=1C=CC=CC=1C(=C(CN)CC)C1=CC=CC=C1 WNKCJOWTKXGERE-UHFFFAOYSA-N 0.000 description 1
- 229950005475 etifelmine Drugs 0.000 description 1
- 229950011633 etifenin Drugs 0.000 description 1
- IBYCYJFUEJQSMK-UHFFFAOYSA-N etifoxine Chemical compound O1C(NCC)=NC2=CC=C(Cl)C=C2C1(C)C1=CC=CC=C1 IBYCYJFUEJQSMK-UHFFFAOYSA-N 0.000 description 1
- 229960003817 etifoxine Drugs 0.000 description 1
- YAGBSNMZQKEFCO-UHFFFAOYSA-N etilamfetamine Chemical compound CCNC(C)CC1=CC=CC=C1 YAGBSNMZQKEFCO-UHFFFAOYSA-N 0.000 description 1
- 229960001003 etilamfetamine Drugs 0.000 description 1
- 229950007285 etintidine Drugs 0.000 description 1
- 229950010483 etipirium iodide Drugs 0.000 description 1
- 229950011035 etiproston Drugs 0.000 description 1
- 229950007353 etiracetam Drugs 0.000 description 1
- LWZCMKGGFONJPB-UHFFFAOYSA-N etiroxate Chemical compound IC1=CC(CC(C)(N)C(=O)OCC)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 LWZCMKGGFONJPB-UHFFFAOYSA-N 0.000 description 1
- 229950010737 etiroxate Drugs 0.000 description 1
- 229950009110 etisazole Drugs 0.000 description 1
- KOBHYGXJFZRQFP-XWXOKSHGSA-N etisomicin Chemical compound O1C[C@](O)(C)[C@@H](NCC)[C@H](O)[C@@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC=C(CN)O2)N)[C@@H](N)C[C@H]1N KOBHYGXJFZRQFP-XWXOKSHGSA-N 0.000 description 1
- 229950007159 etisomicin Drugs 0.000 description 1
- 229950002163 etisulergine Drugs 0.000 description 1
- 229960004404 etizolam Drugs 0.000 description 1
- 229950006579 etocarlide Drugs 0.000 description 1
- 229960005293 etodolac Drugs 0.000 description 1
- XFBVBWWRPKNWHW-UHFFFAOYSA-N etodolac Chemical compound C1COC(CC)(CC(O)=O)C2=N[C]3C(CC)=CC=CC3=C21 XFBVBWWRPKNWHW-UHFFFAOYSA-N 0.000 description 1
- 229960004386 etofamide Drugs 0.000 description 1
- QTRALMGDQMIVFF-UHFFFAOYSA-N etofamide Chemical compound C1=CC(CN(CCOCC)C(=O)C(Cl)Cl)=CC=C1OC1=CC=C([N+]([O-])=O)C=C1 QTRALMGDQMIVFF-UHFFFAOYSA-N 0.000 description 1
- 229960001493 etofenamate Drugs 0.000 description 1
- YREQHYQNNWYQCJ-UHFFFAOYSA-N etofenprox Chemical compound C1=CC(OCC)=CC=C1C(C)(C)COCC1=CC=CC(OC=2C=CC=CC=2)=C1 YREQHYQNNWYQCJ-UHFFFAOYSA-N 0.000 description 1
- 229950005085 etofenprox Drugs 0.000 description 1
- 229960003501 etofibrate Drugs 0.000 description 1
- XXRVYAFBUDSLJX-UHFFFAOYSA-N etofibrate Chemical compound C=1C=CN=CC=1C(=O)OCCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 XXRVYAFBUDSLJX-UHFFFAOYSA-N 0.000 description 1
- 229950000460 etoformin Drugs 0.000 description 1
- 229950002890 etofuradine Drugs 0.000 description 1
- 229960005387 etofylline Drugs 0.000 description 1
- 229960005237 etoglucid Drugs 0.000 description 1
- 229950000181 etolorex Drugs 0.000 description 1
- DDKKBTHTVWQJQX-UHFFFAOYSA-N etolorex Chemical compound OCCNC(C)(C)CC1=CC=C(Cl)C=C1 DDKKBTHTVWQJQX-UHFFFAOYSA-N 0.000 description 1
- 229950000692 etolotifen Drugs 0.000 description 1
- 229950005182 etoloxamine Drugs 0.000 description 1
- 229950004649 etomidoline Drugs 0.000 description 1
- 229950006213 etomoxir Drugs 0.000 description 1
- 229950004538 etonitazene Drugs 0.000 description 1
- PXDBZSCGSQSKST-UHFFFAOYSA-N etonitazene Chemical compound C1=CC(OCC)=CC=C1CC1=NC2=CC([N+]([O-])=O)=CC=C2N1CCN(CC)CC PXDBZSCGSQSKST-UHFFFAOYSA-N 0.000 description 1
- IZBNNCFOBMGTQX-UHFFFAOYSA-N etoperidone Chemical compound O=C1N(CC)C(CC)=NN1CCCN1CCN(C=2C=C(Cl)C=CC=2)CC1 IZBNNCFOBMGTQX-UHFFFAOYSA-N 0.000 description 1
- 229960005437 etoperidone Drugs 0.000 description 1
- 229950000098 etoprindole Drugs 0.000 description 1
- CAHCBJPUTCKATP-FAWZKKEFSA-N etorphine Chemical compound O([C@H]1[C@@]2(OC)C=C[C@@]34C[C@@H]2[C@](C)(O)CCC)C2=C5[C@]41CCN(C)[C@@H]3CC5=CC=C2O CAHCBJPUTCKATP-FAWZKKEFSA-N 0.000 description 1
- 229950004155 etorphine Drugs 0.000 description 1
- 229950008109 etosalamide Drugs 0.000 description 1
- 229950011255 etoxadrol Drugs 0.000 description 1
- INOYCBNLWYEPSB-XHSDSOJGSA-N etoxadrol Chemical compound C([C@H]1[C@H]2CO[C@](O2)(CC)C=2C=CC=CC=2)CCCN1 INOYCBNLWYEPSB-XHSDSOJGSA-N 0.000 description 1
- GAWOVNGQYQVFLI-UHFFFAOYSA-N etoxazene Chemical compound C1=CC(OCC)=CC=C1N=NC1=CC=C(N)C=C1N GAWOVNGQYQVFLI-UHFFFAOYSA-N 0.000 description 1
- 229950008765 etoxazene Drugs 0.000 description 1
- 229950004151 etoxeridine Drugs 0.000 description 1
- 229960004514 etozolin Drugs 0.000 description 1
- ZCKKHYXUQFTBIK-KTKRTIGZSA-N etozoline Chemical compound O=C1N(C)C(=C/C(=O)OCC)/SC1N1CCCCC1 ZCKKHYXUQFTBIK-KTKRTIGZSA-N 0.000 description 1
- 229950008087 etrabamine Drugs 0.000 description 1
- 229960002199 etretinate Drugs 0.000 description 1
- HQMNCQVAMBCHCO-DJRRULDNSA-N etretinate Chemical compound CCOC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)C=C(OC)C(C)=C1C HQMNCQVAMBCHCO-DJRRULDNSA-N 0.000 description 1
- ZXUMUPVQYAFTLF-UHFFFAOYSA-N etryptamine Chemical compound C1=CC=C2C(CC(N)CC)=CNC2=C1 ZXUMUPVQYAFTLF-UHFFFAOYSA-N 0.000 description 1
- 229950005957 etryptamine Drugs 0.000 description 1
- 229950005349 etymemazine Drugs 0.000 description 1
- USKHCLAXJXCWMO-UHFFFAOYSA-N etymemazine Chemical compound C1=CC=C2N(CC(C)CN(C)C)C3=CC(CC)=CC=C3SC2=C1 USKHCLAXJXCWMO-UHFFFAOYSA-N 0.000 description 1
- YGHHWSRCTPQFFC-UHFFFAOYSA-N eucalyptosin A Natural products OC1C(O)C(O)C(CO)OC1OC1C(OC(C#N)C=2C=CC=CC=2)OC(CO)C(O)C1O YGHHWSRCTPQFFC-UHFFFAOYSA-N 0.000 description 1
- 229950002420 eucatropine Drugs 0.000 description 1
- 229960002217 eugenol Drugs 0.000 description 1
- 229950008467 euprocin Drugs 0.000 description 1
- 229950004383 evandamine Drugs 0.000 description 1
- 229960003699 evans blue Drugs 0.000 description 1
- 229950010333 exalamide Drugs 0.000 description 1
- CKSJXOVLXUMMFF-UHFFFAOYSA-N exalamide Chemical compound CCCCCCOC1=CC=CC=C1C(N)=O CKSJXOVLXUMMFF-UHFFFAOYSA-N 0.000 description 1
- ABXHHEZNIJUQFM-UHFFFAOYSA-N exaprolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1C1CCCCC1 ABXHHEZNIJUQFM-UHFFFAOYSA-N 0.000 description 1
- 229950003436 exaprolol Drugs 0.000 description 1
- 229950002388 exepanol Drugs 0.000 description 1
- 229950006404 exifone Drugs 0.000 description 1
- YTPJKQPMTSNTGI-UHFFFAOYSA-N exiproben Chemical compound CCCCCCOCC(O)COC1=CC=CC=C1C(O)=O YTPJKQPMTSNTGI-UHFFFAOYSA-N 0.000 description 1
- 229950006425 exiproben Drugs 0.000 description 1
- 229940012413 factor vii Drugs 0.000 description 1
- 229950001812 falintolol Drugs 0.000 description 1
- 229950001443 falipamil Drugs 0.000 description 1
- UUMGNNQOCVDZDG-UHFFFAOYSA-N falipamil Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCN1C(=O)C2=CC(OC)=C(OC)C=C2C1 UUMGNNQOCVDZDG-UHFFFAOYSA-N 0.000 description 1
- 229950007632 famiraprinium chloride Drugs 0.000 description 1
- 229960001596 famotidine Drugs 0.000 description 1
- XUFQPHANEAPEMJ-UHFFFAOYSA-N famotidine Chemical compound NC(N)=NC1=NC(CSCCC(N)=NS(N)(=O)=O)=CS1 XUFQPHANEAPEMJ-UHFFFAOYSA-N 0.000 description 1
- 229950001806 famotine Drugs 0.000 description 1
- 229950004429 fanetizole Drugs 0.000 description 1
- WEEYMMXMBFJUAI-UHFFFAOYSA-N fanetizole Chemical compound N=1C(C=2C=CC=CC=2)=CSC=1NCCC1=CC=CC=C1 WEEYMMXMBFJUAI-UHFFFAOYSA-N 0.000 description 1
- 229950011619 fantridone Drugs 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 229960000546 fazadinium bromide Drugs 0.000 description 1
- 229960005282 febantel Drugs 0.000 description 1
- QHZQILHUJDRDAI-UHFFFAOYSA-N febarbamate Chemical compound O=C1N(CC(COCCCC)OC(N)=O)C(=O)NC(=O)C1(CC)C1=CC=CC=C1 QHZQILHUJDRDAI-UHFFFAOYSA-N 0.000 description 1
- 229960005182 febarbamate Drugs 0.000 description 1
- 229950002622 febuverine Drugs 0.000 description 1
- 229950001582 feclemine Drugs 0.000 description 1
- QDMORDTWFMWOFA-UHFFFAOYSA-N feclemine Chemical compound C=1C=CC=CC=1C(C(CN(CC)CC)CN(CC)CC)C1CCCCC1 QDMORDTWFMWOFA-UHFFFAOYSA-N 0.000 description 1
- OZKQTMYKYQGCME-UHFFFAOYSA-N feclobuzone Chemical compound O=C1N(C=2C=CC=CC=2)N(C=2C=CC=CC=2)C(=O)C1(CCCC)COC(=O)C1=CC=C(Cl)C=C1 OZKQTMYKYQGCME-UHFFFAOYSA-N 0.000 description 1
- 229950004534 feclobuzone Drugs 0.000 description 1
- 229960004049 fedrilate Drugs 0.000 description 1
- RDEOYUSTRWNWLX-UHFFFAOYSA-N fedrilate Chemical compound C1COCCC1(C=1C=CC=CC=1)C(=O)OC(C)CCN1CCOCC1 RDEOYUSTRWNWLX-UHFFFAOYSA-N 0.000 description 1
- 229960003472 felbamate Drugs 0.000 description 1
- WKGXYQFOCVYPAC-UHFFFAOYSA-N felbamate Chemical compound NC(=O)OCC(COC(N)=O)C1=CC=CC=C1 WKGXYQFOCVYPAC-UHFFFAOYSA-N 0.000 description 1
- 229960000192 felbinac Drugs 0.000 description 1
- 229950000897 felipyrine Drugs 0.000 description 1
- 229960003580 felodipine Drugs 0.000 description 1
- 229950003930 femoxetine Drugs 0.000 description 1
- OJSFTALXCYKKFQ-YLJYHZDGSA-N femoxetine Chemical compound C1=CC(OC)=CC=C1OC[C@@H]1[C@@H](C=2C=CC=CC=2)CCN(C)C1 OJSFTALXCYKKFQ-YLJYHZDGSA-N 0.000 description 1
- 229950002349 fenabutene Drugs 0.000 description 1
- 229950005064 fenacetinol Drugs 0.000 description 1
- CFLMLQJYYALAPD-UHFFFAOYSA-N fenacetinol Natural products CCOC1=CC=C(NC(C)=O)C(O)=C1 CFLMLQJYYALAPD-UHFFFAOYSA-N 0.000 description 1
- 229950006308 fenaclon Drugs 0.000 description 1
- OINXXHYENYVYPB-UHFFFAOYSA-N fenadiazole Chemical compound OC1=CC=CC=C1C1=NN=CO1 OINXXHYENYVYPB-UHFFFAOYSA-N 0.000 description 1
- 229950001092 fenadiazole Drugs 0.000 description 1
- RMQKPRRJSKFBRU-UHFFFAOYSA-N fenalamide Chemical compound CCN(CC)CCNC(=O)C(CC)(C(=O)OCC)C1=CC=CC=C1 RMQKPRRJSKFBRU-UHFFFAOYSA-N 0.000 description 1
- 229950006906 fenalamide Drugs 0.000 description 1
- DOBLSWXRNYSVDC-UHFFFAOYSA-N fenalcomine Chemical compound C1=CC(C(O)CC)=CC=C1OCCNC(C)CC1=CC=CC=C1 DOBLSWXRNYSVDC-UHFFFAOYSA-N 0.000 description 1
- 229950009129 fenalcomine Drugs 0.000 description 1
- 229950002788 fenamifuril Drugs 0.000 description 1
- 229950003495 fenaperone Drugs 0.000 description 1
- 229960005473 fenbendazole Drugs 0.000 description 1
- IRHZVMHXVHSMKB-UHFFFAOYSA-N fenbendazole Chemical compound [CH]1C2=NC(NC(=O)OC)=NC2=CC=C1SC1=CC=CC=C1 IRHZVMHXVHSMKB-UHFFFAOYSA-N 0.000 description 1
- 229960001395 fenbufen Drugs 0.000 description 1
- ZPAKPRAICRBAOD-UHFFFAOYSA-N fenbufen Chemical compound C1=CC(C(=O)CCC(=O)O)=CC=C1C1=CC=CC=C1 ZPAKPRAICRBAOD-UHFFFAOYSA-N 0.000 description 1
- 229960002533 fenbutrazate Drugs 0.000 description 1
- BAQKJENAVQLANS-UHFFFAOYSA-N fenbutrazate Chemical compound C=1C=CC=CC=1C(CC)C(=O)OCCN(C1C)CCOC1C1=CC=CC=C1 BAQKJENAVQLANS-UHFFFAOYSA-N 0.000 description 1
- 229960001938 fencamfamin Drugs 0.000 description 1
- IKFBPFGUINLYQI-UHFFFAOYSA-N fencamfamin Chemical compound CCNC1C(C2)CCC2C1C1=CC=CC=C1 IKFBPFGUINLYQI-UHFFFAOYSA-N 0.000 description 1
- 229950000795 fencibutirol Drugs 0.000 description 1
- HPLFQKUIHGZHPH-UHFFFAOYSA-N fencibutirol Chemical compound C1CC(C(C(O)=O)CC)(O)CCC1C1=CC=CC=C1 HPLFQKUIHGZHPH-UHFFFAOYSA-N 0.000 description 1
- 229950002164 fenclexonium metilsulfate Drugs 0.000 description 1
- 229950003853 fenclonine Drugs 0.000 description 1
- 229950003537 fenclorac Drugs 0.000 description 1
- 229960002602 fendiline Drugs 0.000 description 1
- HAWWPSYXSLJRBO-UHFFFAOYSA-N fendosal Chemical compound C1=C(O)C(C(=O)O)=CC(N2C(=CC=3C4=CC=CC=C4CCC=32)C=2C=CC=CC=2)=C1 HAWWPSYXSLJRBO-UHFFFAOYSA-N 0.000 description 1
- 229950005416 fendosal Drugs 0.000 description 1
- 229950008296 feneritrol Drugs 0.000 description 1
- 229950004104 fenestrel Drugs 0.000 description 1
- 229950007454 fenethazine Drugs 0.000 description 1
- PFAXACNYGZVKMX-UHFFFAOYSA-N fenethazine Chemical compound C1=CC=C2N(CCN(C)C)C3=CC=CC=C3SC2=C1 PFAXACNYGZVKMX-UHFFFAOYSA-N 0.000 description 1
- 229940032465 fenethylline Drugs 0.000 description 1
- 229950001309 fenetradil Drugs 0.000 description 1
- ITFWPRPSIAYKMV-UHFFFAOYSA-N fenflumizol Chemical compound C1=CC(OC)=CC=C1C1=C(C=2C=CC(OC)=CC=2)NC(C=2C(=CC(F)=CC=2)F)=N1 ITFWPRPSIAYKMV-UHFFFAOYSA-N 0.000 description 1
- 229960001582 fenfluramine Drugs 0.000 description 1
- 229950006668 fenfluthrin Drugs 0.000 description 1
- 229950004395 fengabine Drugs 0.000 description 1
- 229950003393 fenharmane Drugs 0.000 description 1
- 229950011506 fenimide Drugs 0.000 description 1
- 229950005975 feniodium chloride Drugs 0.000 description 1
- 229960005035 fenipentol Drugs 0.000 description 1
- 229950007132 fenirofibrate Drugs 0.000 description 1
- 229950000734 fenisorex Drugs 0.000 description 1
- HEXAHJRXDZDVLR-HZPDHXFCSA-N fenisorex Chemical compound C1([C@@H]2C3=CC(F)=CC=C3C[C@@H](O2)NC)=CC=CC=C1 HEXAHJRXDZDVLR-HZPDHXFCSA-N 0.000 description 1
- FVHAONUKSUFJKN-UHFFFAOYSA-N fenmetozole Chemical compound C1=C(Cl)C(Cl)=CC=C1OCC1=NCCN1 FVHAONUKSUFJKN-UHFFFAOYSA-N 0.000 description 1
- 229950009662 fenmetozole Drugs 0.000 description 1
- 229950011062 fenmetramide Drugs 0.000 description 1
- 229950002489 fenobam Drugs 0.000 description 1
- 229950001026 fenocinol Drugs 0.000 description 1
- 229950010805 fenoctimine Drugs 0.000 description 1
- 229960002297 fenofibrate Drugs 0.000 description 1
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 1
- 229960002724 fenoldopam Drugs 0.000 description 1
- TVURRHSHRRELCG-UHFFFAOYSA-N fenoldopam Chemical compound C1=CC(O)=CC=C1C1C2=CC(O)=C(O)C(Cl)=C2CCNC1 TVURRHSHRRELCG-UHFFFAOYSA-N 0.000 description 1
- 229960001419 fenoprofen Drugs 0.000 description 1
- 229960001022 fenoterol Drugs 0.000 description 1
- UBAJTZKNDCEGKL-UHFFFAOYSA-N fenoverine Chemical compound C12=CC=CC=C2SC2=CC=CC=C2N1C(=O)CN1CCN(CC=2C=C3OCOC3=CC=2)CC1 UBAJTZKNDCEGKL-UHFFFAOYSA-N 0.000 description 1
- 229960001966 fenoverine Drugs 0.000 description 1
- 229960004970 fenoxazoline Drugs 0.000 description 1
- GFYSWQDCHLWRMQ-UHFFFAOYSA-N fenoxazoline Chemical compound CC(C)C1=CC=CC=C1OCC1=NCCN1 GFYSWQDCHLWRMQ-UHFFFAOYSA-N 0.000 description 1
- OBQUKWIVMOIRGG-UHFFFAOYSA-N fenoxedil Chemical compound C1=CC(OCCCC)=CC=C1OCC(=O)N(CCN(CC)CC)C1=CC(OCC)=CC=C1OCC OBQUKWIVMOIRGG-UHFFFAOYSA-N 0.000 description 1
- 229950011050 fenoxedil Drugs 0.000 description 1
- 229960001038 fenozolone Drugs 0.000 description 1
- RXOIEVSUURELPG-UHFFFAOYSA-N fenozolone Chemical compound O1C(NCC)=NC(=O)C1C1=CC=CC=C1 RXOIEVSUURELPG-UHFFFAOYSA-N 0.000 description 1
- 229950011196 fenpentadiol Drugs 0.000 description 1
- SNJDSTGQYRTZJT-UHFFFAOYSA-N fenpentadiol Chemical compound CC(C)(O)CC(C)(O)C1=CC=C(Cl)C=C1 SNJDSTGQYRTZJT-UHFFFAOYSA-N 0.000 description 1
- 229950004379 fenperate Drugs 0.000 description 1
- 229950005166 fenpiverinium bromide Drugs 0.000 description 1
- 229950008746 fenprinast Drugs 0.000 description 1
- IQUFSXIQAFPIMR-UHFFFAOYSA-N fenproporex Chemical compound N#CCCNC(C)CC1=CC=CC=C1 IQUFSXIQAFPIMR-UHFFFAOYSA-N 0.000 description 1
- 229960005231 fenproporex Drugs 0.000 description 1
- 229950003109 fenprostalene Drugs 0.000 description 1
- 229960002637 fenquizone Drugs 0.000 description 1
- DBDTUXMDTSTPQZ-UHFFFAOYSA-N fenquizone Chemical compound N1C=2C=C(Cl)C(S(=O)(=O)N)=CC=2C(=O)NC1C1=CC=CC=C1 DBDTUXMDTSTPQZ-UHFFFAOYSA-N 0.000 description 1
- 229950003662 fenretinide Drugs 0.000 description 1
- 229960002912 fenspiride Drugs 0.000 description 1
- 229960002428 fentanyl Drugs 0.000 description 1
- 229960004207 fentanyl citrate Drugs 0.000 description 1
- 229960002679 fentiazac Drugs 0.000 description 1
- 229950005344 fenticlor Drugs 0.000 description 1
- ANUSOIHIIPAHJV-UHFFFAOYSA-N fenticlor Chemical compound OC1=CC=C(Cl)C=C1SC1=CC(Cl)=CC=C1O ANUSOIHIIPAHJV-UHFFFAOYSA-N 0.000 description 1
- 229960001274 fenticonazole Drugs 0.000 description 1
- 229950002909 fentonium bromide Drugs 0.000 description 1
- 229950003541 fenyripol Drugs 0.000 description 1
- 229950011603 fepentolic acid Drugs 0.000 description 1
- 229950004007 fepitrizol Drugs 0.000 description 1
- PVOOBRUZWPQOER-UHFFFAOYSA-N fepradinol Chemical compound OCC(C)(C)NCC(O)C1=CC=CC=C1 PVOOBRUZWPQOER-UHFFFAOYSA-N 0.000 description 1
- 229950008205 fepradinol Drugs 0.000 description 1
- 229960000489 feprazone Drugs 0.000 description 1
- 229950004190 fepromide Drugs 0.000 description 1
- 229950002525 feprosidnine Drugs 0.000 description 1
- HFLCEELTJROKMJ-UHFFFAOYSA-N feprosidnine Chemical compound C1=C([NH-])ON=[N+]1C(C)CC1=CC=CC=C1 HFLCEELTJROKMJ-UHFFFAOYSA-N 0.000 description 1
- 229950002211 ferrotrenine Drugs 0.000 description 1
- 239000011773 ferrous fumarate Substances 0.000 description 1
- 235000002332 ferrous fumarate Nutrition 0.000 description 1
- 229960000225 ferrous fumarate Drugs 0.000 description 1
- 235000013924 ferrous gluconate Nutrition 0.000 description 1
- 239000004222 ferrous gluconate Substances 0.000 description 1
- 229960001645 ferrous gluconate Drugs 0.000 description 1
- 229950009523 fexicaine Drugs 0.000 description 1
- MIWWSGDADVMLTG-UHFFFAOYSA-N fexinidazole Chemical compound C1=CC(SC)=CC=C1OCC1=NC=C([N+]([O-])=O)N1C MIWWSGDADVMLTG-UHFFFAOYSA-N 0.000 description 1
- 229950004464 fexinidazole Drugs 0.000 description 1
- 229950002557 fezatione Drugs 0.000 description 1
- NELSQLPTEWCHQW-UHFFFAOYSA-N fezolamine Chemical compound N=1N(CCCN(C)C)C=C(C=2C=CC=CC=2)C=1C1=CC=CC=C1 NELSQLPTEWCHQW-UHFFFAOYSA-N 0.000 description 1
- 229950000761 fezolamine Drugs 0.000 description 1
- 229950003564 fiacitabine Drugs 0.000 description 1
- 229950006401 fibracillin Drugs 0.000 description 1
- 108010020275 fibrin receptor Proteins 0.000 description 1
- 239000003527 fibrinolytic agent Substances 0.000 description 1
- 102000036072 fibronectin binding proteins Human genes 0.000 description 1
- 108091010988 fibronectin binding proteins Proteins 0.000 description 1
- KFSXLIJSXOJBCB-HZMBPMFUSA-N filenadol Chemical compound N1([C@@H]([C@@H](O)C)C=2C=C3OCOC3=CC=2)CCOCC1 KFSXLIJSXOJBCB-HZMBPMFUSA-N 0.000 description 1
- 229950003056 filenadol Drugs 0.000 description 1
- IMQSIXYSKPIGPD-NKYUYKLDSA-N filipin Chemical compound CCCCC[C@H](O)[C@@H]1[C@@H](O)C[C@@H](O)C[C@@H](O)C[C@@H](O)C[C@@H](O)C[C@@H](O)C[C@H](O)\C(C)=C\C=C\C=C\C=C\C=C\[C@H](O)[C@@H](C)OC1=O IMQSIXYSKPIGPD-NKYUYKLDSA-N 0.000 description 1
- 229950000152 filipin Drugs 0.000 description 1
- IMQSIXYSKPIGPD-UHFFFAOYSA-N filipin III Natural products CCCCCC(O)C1C(O)CC(O)CC(O)CC(O)CC(O)CC(O)CC(O)C(C)=CC=CC=CC=CC=CC(O)C(C)OC1=O IMQSIXYSKPIGPD-UHFFFAOYSA-N 0.000 description 1
- HDVRLUFGYQYLFJ-UHFFFAOYSA-N flamenol Chemical compound COC1=CC(O)=CC(O)=C1 HDVRLUFGYQYLFJ-UHFFFAOYSA-N 0.000 description 1
- 229950001729 flamenol Drugs 0.000 description 1
- 229950010661 flavamine Drugs 0.000 description 1
- IGCSSLDDCHLXGL-UHFFFAOYSA-N flavodic acid Chemical compound C=1C(OCC(=O)O)=CC(OCC(O)=O)=C(C(C=2)=O)C=1OC=2C1=CC=CC=C1 IGCSSLDDCHLXGL-UHFFFAOYSA-N 0.000 description 1
- 229960005043 flavodic acid Drugs 0.000 description 1
- 229960000855 flavoxate Drugs 0.000 description 1
- SPIUTQOUKAMGCX-UHFFFAOYSA-N flavoxate Chemical compound C1=CC=C2C(=O)C(C)=C(C=3C=CC=CC=3)OC2=C1C(=O)OCCN1CCCCC1 SPIUTQOUKAMGCX-UHFFFAOYSA-N 0.000 description 1
- 229950004322 flazalone Drugs 0.000 description 1
- 229960000449 flecainide Drugs 0.000 description 1
- 229950009061 flerobuterol Drugs 0.000 description 1
- 229960003306 fleroxacin Drugs 0.000 description 1
- XBJBPGROQZJDOJ-UHFFFAOYSA-N fleroxacin Chemical compound C1CN(C)CCN1C1=C(F)C=C2C(=O)C(C(O)=O)=CN(CCF)C2=C1F XBJBPGROQZJDOJ-UHFFFAOYSA-N 0.000 description 1
- NYSDRDDQELAVKP-SFHVURJKSA-N flesinoxan Chemical compound C([C@@H](O1)CO)OC2=C1C=CC=C2N(CC1)CCN1CCNC(=O)C1=CC=C(F)C=C1 NYSDRDDQELAVKP-SFHVURJKSA-N 0.000 description 1
- 229950003678 flesinoxan Drugs 0.000 description 1
- ZPLOQFLCMVIWRY-UHFFFAOYSA-N flestolol Chemical compound NC(=O)NCC(C)(C)NCC(O)COC(=O)C1=CC=CC=C1F ZPLOQFLCMVIWRY-UHFFFAOYSA-N 0.000 description 1
- 229950001315 flestolol Drugs 0.000 description 1
- CIZCSUNUBQXVFP-UHFFFAOYSA-N fletazepam Chemical compound FC1=CC=CC=C1C1=NCCN(CC(F)(F)F)C2=CC=C(Cl)C=C12 CIZCSUNUBQXVFP-UHFFFAOYSA-N 0.000 description 1
- 229950005700 fletazepam Drugs 0.000 description 1
- 229960003240 floctafenine Drugs 0.000 description 1
- 229960002878 flomoxef Drugs 0.000 description 1
- UHRBTBZOWWGKMK-DOMZBBRYSA-N flomoxef Chemical compound O([C@@H]1[C@@](C(N1C=1C(O)=O)=O)(NC(=O)CSC(F)F)OC)CC=1CSC1=NN=NN1CCO UHRBTBZOWWGKMK-DOMZBBRYSA-N 0.000 description 1
- 229960000430 flopropione Drugs 0.000 description 1
- 229950005493 florantyrone Drugs 0.000 description 1
- QOBAOSCOLAGPKI-UHFFFAOYSA-N florantyrone Chemical compound C1=CC(C2=CC=C(C=C22)C(=O)CCC(=O)O)=C3C2=CC=CC3=C1 QOBAOSCOLAGPKI-UHFFFAOYSA-N 0.000 description 1
- RSXGUJLKWYUPMC-UHFFFAOYSA-N flordipine Chemical compound CC1=C(C(=O)OCC)C(C=2C(=CC=CC=2)C(F)(F)F)C(C(=O)OCC)=C(C)N1CCN1CCOCC1 RSXGUJLKWYUPMC-UHFFFAOYSA-N 0.000 description 1
- 229950009366 flordipine Drugs 0.000 description 1
- 229950011336 floredil Drugs 0.000 description 1
- MXVLJFCCQMXEEE-UHFFFAOYSA-N floredil Chemical compound CCOC1=CC(OCC)=CC(OCCN2CCOCC2)=C1 MXVLJFCCQMXEEE-UHFFFAOYSA-N 0.000 description 1
- 229960003760 florfenicol Drugs 0.000 description 1
- 229950002994 florifenine Drugs 0.000 description 1
- 229960001606 flosequinan Drugs 0.000 description 1
- UYGONJYYUKVHDD-UHFFFAOYSA-N flosequinan Chemical compound C1=C(F)C=C2N(C)C=C(S(C)=O)C(=O)C2=C1 UYGONJYYUKVHDD-UHFFFAOYSA-N 0.000 description 1
- 229950003519 flotrenizine Drugs 0.000 description 1
- 229950003049 floverine Drugs 0.000 description 1
- 229960004273 floxacillin Drugs 0.000 description 1
- 229950002996 floxacrine Drugs 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 229950002413 fluacizine Drugs 0.000 description 1
- VHEOUJNDDFHPGJ-UHFFFAOYSA-N fluacizine Chemical compound C1=C(C(F)(F)F)C=C2N(C(=O)CCN(CC)CC)C3=CC=CC=C3SC2=C1 VHEOUJNDDFHPGJ-UHFFFAOYSA-N 0.000 description 1
- 229950007846 flualamide Drugs 0.000 description 1
- 229960005220 fluanisone Drugs 0.000 description 1
- IRYFCWPNDIUQOW-UHFFFAOYSA-N fluanisone Chemical compound COC1=CC=CC=C1N1CCN(CCCC(=O)C=2C=CC(F)=CC=2)CC1 IRYFCWPNDIUQOW-UHFFFAOYSA-N 0.000 description 1
- BYZCJOHDXLROEC-RBWIMXSLSA-N fluazacort Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)=N[C@@]3(C(=O)COC(=O)C)[C@@]1(C)C[C@@H]2O BYZCJOHDXLROEC-RBWIMXSLSA-N 0.000 description 1
- 229950002335 fluazacort Drugs 0.000 description 1
- 229950003191 flubanilate Drugs 0.000 description 1
- 229960004500 flubendazole Drugs 0.000 description 1
- CPEUVMUXAHMANV-UHFFFAOYSA-N flubendazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=C(F)C=C1 CPEUVMUXAHMANV-UHFFFAOYSA-N 0.000 description 1
- 229950005430 flubepride Drugs 0.000 description 1
- SKWBJBJQEUEFCY-UHFFFAOYSA-N flucetorex Chemical compound C=1C=C(NC(C)=O)C=CC=1OCC(=O)NC(C)CC1=CC=CC(C(F)(F)F)=C1 SKWBJBJQEUEFCY-UHFFFAOYSA-N 0.000 description 1
- 229950003524 flucetorex Drugs 0.000 description 1
- 229950001600 flucindole Drugs 0.000 description 1
- FXNCRITWFOVSEP-UHFFFAOYSA-N flucindole Chemical compound N1C2=C(F)C=C(F)C=C2C2=C1CCC(N(C)C)C2 FXNCRITWFOVSEP-UHFFFAOYSA-N 0.000 description 1
- 229950003881 fluciprazine Drugs 0.000 description 1
- NJNWEGFJCGYWQT-VSXGLTOVSA-N fluclorolone acetonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(Cl)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1Cl NJNWEGFJCGYWQT-VSXGLTOVSA-N 0.000 description 1
- 229940094766 flucloronide Drugs 0.000 description 1
- 229960004884 fluconazole Drugs 0.000 description 1
- RFHAOTPXVQNOHP-UHFFFAOYSA-N fluconazole Chemical compound C1=NC=NN1CC(C=1C(=CC(F)=CC=1)F)(O)CN1C=NC=N1 RFHAOTPXVQNOHP-UHFFFAOYSA-N 0.000 description 1
- 229950009047 fludalanine Drugs 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 229950002840 fludazonium chloride Drugs 0.000 description 1
- 229960004930 fludiazepam Drugs 0.000 description 1
- ROYOYTLGDLIGBX-UHFFFAOYSA-N fludiazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F ROYOYTLGDLIGBX-UHFFFAOYSA-N 0.000 description 1
- 229950002723 fludorex Drugs 0.000 description 1
- 229950000994 fludoxopone Drugs 0.000 description 1
- 229960004369 flufenamic acid Drugs 0.000 description 1
- LPEPZBJOKDYZAD-UHFFFAOYSA-N flufenamic acid Chemical compound OC(=O)C1=CC=CC=C1NC1=CC=CC(C(F)(F)F)=C1 LPEPZBJOKDYZAD-UHFFFAOYSA-N 0.000 description 1
- 229950007979 flufenisal Drugs 0.000 description 1
- 229950009037 flufosal Drugs 0.000 description 1
- 229950006968 flufylline Drugs 0.000 description 1
- 229950010023 fluindarol Drugs 0.000 description 1
- 229960005298 fluindione Drugs 0.000 description 1
- NASXCEITKQITLD-UHFFFAOYSA-N fluindione Chemical compound C1=CC(F)=CC=C1C1C(=O)C2=CC=CC=C2C1=O NASXCEITKQITLD-UHFFFAOYSA-N 0.000 description 1
- 229960004381 flumazenil Drugs 0.000 description 1
- OFBIFZUFASYYRE-UHFFFAOYSA-N flumazenil Chemical compound C1N(C)C(=O)C2=CC(F)=CC=C2N2C=NC(C(=O)OCC)=C21 OFBIFZUFASYYRE-UHFFFAOYSA-N 0.000 description 1
- 229950004612 flumecinol Drugs 0.000 description 1
- DVASNQYQOZHAJN-UHFFFAOYSA-N flumecinol Chemical compound C=1C=CC(C(F)(F)F)=CC=1C(O)(CC)C1=CC=CC=C1 DVASNQYQOZHAJN-UHFFFAOYSA-N 0.000 description 1
- 229960000702 flumequine Drugs 0.000 description 1
- 229950002450 flumeridone Drugs 0.000 description 1
- 229960003469 flumetasone Drugs 0.000 description 1
- WXURHACBFYSXBI-GQKYHHCASA-N flumethasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]2(C)C[C@@H]1O WXURHACBFYSXBI-GQKYHHCASA-N 0.000 description 1
- 229940042902 flumethasone pivalate Drugs 0.000 description 1
- JWRMHDSINXPDHB-OJAGFMMFSA-N flumethasone pivalate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)COC(=O)C(C)(C)C)(O)[C@@]2(C)C[C@@H]1O JWRMHDSINXPDHB-OJAGFMMFSA-N 0.000 description 1
- 229960003028 flumethiazide Drugs 0.000 description 1
- RGUQWGXAYZNLMI-UHFFFAOYSA-N flumethiazide Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC2=C1NC=NS2(=O)=O RGUQWGXAYZNLMI-UHFFFAOYSA-N 0.000 description 1
- 229950006843 flumetramide Drugs 0.000 description 1
- 229950010839 flumexadol Drugs 0.000 description 1
- 229950005785 flumezapine Drugs 0.000 description 1
- NMGYDYBWRZHLHR-UHFFFAOYSA-N fluminorex Chemical compound O1C(N)=NCC1C1=CC=C(C(F)(F)F)C=C1 NMGYDYBWRZHLHR-UHFFFAOYSA-N 0.000 description 1
- 229950007852 fluminorex Drugs 0.000 description 1
- OPYFPDBMMYUPME-UHFFFAOYSA-N flumizole Chemical compound C1=CC(OC)=CC=C1C1=C(C=2C=CC(OC)=CC=2)NC(C(F)(F)F)=N1 OPYFPDBMMYUPME-UHFFFAOYSA-N 0.000 description 1
- 229950005288 flumizole Drugs 0.000 description 1
- 229950002998 flumoxonide Drugs 0.000 description 1
- 229950004385 flunamine Drugs 0.000 description 1
- SMANXXCATUTDDT-QPJJXVBHSA-N flunarizine Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)N1CCN(C\C=C\C=2C=CC=CC=2)CC1 SMANXXCATUTDDT-QPJJXVBHSA-N 0.000 description 1
- 229960000326 flunarizine Drugs 0.000 description 1
- 229950010177 flunidazole Drugs 0.000 description 1
- WEGNFRKBIKYVLC-XTLNBZDDSA-N flunisolide acetate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)COC(=O)C)[C@@]2(C)C[C@@H]1O WEGNFRKBIKYVLC-XTLNBZDDSA-N 0.000 description 1
- 229960000588 flunixin Drugs 0.000 description 1
- NOOCSNJCXJYGPE-UHFFFAOYSA-N flunixin Chemical compound C1=CC=C(C(F)(F)F)C(C)=C1NC1=NC=CC=C1C(O)=O NOOCSNJCXJYGPE-UHFFFAOYSA-N 0.000 description 1
- 229950006292 flunoprost Drugs 0.000 description 1
- 229960001321 flunoxaprofen Drugs 0.000 description 1
- ARPYQKTVRGFPIS-VIFPVBQESA-N flunoxaprofen Chemical compound N=1C2=CC([C@@H](C(O)=O)C)=CC=C2OC=1C1=CC=C(F)C=C1 ARPYQKTVRGFPIS-VIFPVBQESA-N 0.000 description 1
- 229960001347 fluocinolone acetonide Drugs 0.000 description 1
- FEBLZLNTKCEFIT-VSXGLTOVSA-N fluocinolone acetonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O FEBLZLNTKCEFIT-VSXGLTOVSA-N 0.000 description 1
- 229960000785 fluocinonide Drugs 0.000 description 1
- GAKMQHDJQHZUTJ-ULHLPKEOSA-N fluocortolone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)CO)[C@@]2(C)C[C@@H]1O GAKMQHDJQHZUTJ-ULHLPKEOSA-N 0.000 description 1
- 229960003973 fluocortolone Drugs 0.000 description 1
- 229960004437 fluocortolone caproate Drugs 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 229960002143 fluorescein Drugs 0.000 description 1
- PRNNIHPVNFPWAH-UHFFFAOYSA-N fluoresone Chemical compound CCS(=O)(=O)C1=CC=C(F)C=C1 PRNNIHPVNFPWAH-UHFFFAOYSA-N 0.000 description 1
- 229950011300 fluoresone Drugs 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- FAOZLTXFLGPHNG-KNAQIMQKSA-N fluorometholone Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@]2(F)[C@@H](O)C[C@]2(C)[C@@](O)(C(C)=O)CC[C@H]21 FAOZLTXFLGPHNG-KNAQIMQKSA-N 0.000 description 1
- 229960001048 fluorometholone Drugs 0.000 description 1
- YRFXGQHBPBMFHW-SBTZIJSASA-N fluorometholone acetate Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@]2(F)[C@@H](O)C[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 YRFXGQHBPBMFHW-SBTZIJSASA-N 0.000 description 1
- 229960001629 fluorometholone acetate Drugs 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 229950006420 fluotracen Drugs 0.000 description 1
- 229960002464 fluoxetine Drugs 0.000 description 1
- 229960001751 fluoxymesterone Drugs 0.000 description 1
- YLRFCQOZQXIBAB-RBZZARIASA-N fluoxymesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@@H]2O YLRFCQOZQXIBAB-RBZZARIASA-N 0.000 description 1
- 229950006702 fluparoxan Drugs 0.000 description 1
- XSOUHEXVEOQRKJ-IUCAKERBSA-N fluparoxan Chemical compound O1[C@H]2CNC[C@@H]2OC2=C1C=CC=C2F XSOUHEXVEOQRKJ-IUCAKERBSA-N 0.000 description 1
- 229960002419 flupentixol Drugs 0.000 description 1
- 229950005771 fluperamide Drugs 0.000 description 1
- 229950010896 fluperlapine Drugs 0.000 description 1
- 229960003590 fluperolone Drugs 0.000 description 1
- HHPZZKDXAFJLOH-QZIXMDIESA-N fluperolone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@@](C(=O)[C@@H](OC(C)=O)C)(O)[C@@]1(C)C[C@@H]2O HHPZZKDXAFJLOH-QZIXMDIESA-N 0.000 description 1
- 229960002690 fluphenazine Drugs 0.000 description 1
- 229960000787 fluphenazine enanthate Drugs 0.000 description 1
- 229950002254 flupimazine Drugs 0.000 description 1
- 229960003667 flupirtine Drugs 0.000 description 1
- 229950002578 flupranone Drugs 0.000 description 1
- 229950006789 fluprazine Drugs 0.000 description 1
- CHQKHXJPWDSTPY-UHFFFAOYSA-N fluprazine Chemical compound C1CN(CCNC(=O)N)CCN1C1=CC=CC(C(F)(F)F)=C1 CHQKHXJPWDSTPY-UHFFFAOYSA-N 0.000 description 1
- 229960003238 fluprednidene Drugs 0.000 description 1
- YVHXHNGGPURVOS-SBTDHBFYSA-N fluprednidene Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@](C(=C)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 YVHXHNGGPURVOS-SBTDHBFYSA-N 0.000 description 1
- 229960000618 fluprednisolone Drugs 0.000 description 1
- 229950001284 fluprofen Drugs 0.000 description 1
- 229950006834 fluprofylline Drugs 0.000 description 1
- ZWOUXWWGKJBAHQ-UHFFFAOYSA-N fluproquazone Chemical compound N=1C(=O)N(C(C)C)C2=CC(C)=CC=C2C=1C1=CC=C(F)C=C1 ZWOUXWWGKJBAHQ-UHFFFAOYSA-N 0.000 description 1
- 229950004250 fluproquazone Drugs 0.000 description 1
- WWSWYXNVCBLWNZ-QIZQQNKQSA-N fluprostenol Chemical compound C([C@H](O)\C=C\[C@@H]1[C@H]([C@@H](O)C[C@H]1O)C\C=C/CCCC(O)=O)OC1=CC=CC(C(F)(F)F)=C1 WWSWYXNVCBLWNZ-QIZQQNKQSA-N 0.000 description 1
- 229950009951 fluprostenol Drugs 0.000 description 1
- 229950007253 fluquazone Drugs 0.000 description 1
- 229950007978 fluradoline Drugs 0.000 description 1
- 229950001572 flurantel Drugs 0.000 description 1
- 229960003528 flurazepam Drugs 0.000 description 1
- 229960003628 flurazepam hydrochloride Drugs 0.000 description 1
- 229960002390 flurbiprofen Drugs 0.000 description 1
- SYTBZMRGLBWNTM-UHFFFAOYSA-N flurbiprofen Chemical compound FC1=CC(C(C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-UHFFFAOYSA-N 0.000 description 1
- 229950003750 fluretofen Drugs 0.000 description 1
- 229960001398 flurithromycin Drugs 0.000 description 1
- XOEUHCONYHZURQ-HNUBZJOYSA-N flurithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@@](C)(F)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 XOEUHCONYHZURQ-HNUBZJOYSA-N 0.000 description 1
- 229950005682 flurocitabine Drugs 0.000 description 1
- 229950007143 flurofamide Drugs 0.000 description 1
- JKQQZJHNUVDHKP-SZMVRVGJSA-N flurogestone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@]2(F)[C@H]1[C@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@]1(C)C[C@@H]2O JKQQZJHNUVDHKP-SZMVRVGJSA-N 0.000 description 1
- KGPPDNUWZNWPSI-UHFFFAOYSA-N flurotyl Chemical group FC(F)(F)COCC(F)(F)F KGPPDNUWZNWPSI-UHFFFAOYSA-N 0.000 description 1
- 229950000929 flurotyl Drugs 0.000 description 1
- DLEGDLSLRSOURQ-UHFFFAOYSA-N fluroxene Chemical compound FC(F)(F)COC=C DLEGDLSLRSOURQ-UHFFFAOYSA-N 0.000 description 1
- 229950010045 fluroxene Drugs 0.000 description 1
- VYKKDKFTDMVOBU-UHFFFAOYSA-N flusalan Chemical compound OC1=C(Br)C=C(Br)C=C1C(=O)NC1=CC=CC(C(F)(F)F)=C1 VYKKDKFTDMVOBU-UHFFFAOYSA-N 0.000 description 1
- 229950004696 flusalan Drugs 0.000 description 1
- 229950010665 flusoxolol Drugs 0.000 description 1
- CYCXZGUVPDRYLG-HXUWFJFHSA-N flusoxolol Chemical compound C1=CC(OC[C@H](O)CNC(C)C)=CC=C1OCCOCCC1=CC=C(F)C=C1 CYCXZGUVPDRYLG-HXUWFJFHSA-N 0.000 description 1
- RIOZXKPJYKSKJV-UHFFFAOYSA-N fluspiperone Chemical compound C1=CC(F)=CC=C1N1C2(CCN(CCCC(=O)C=3C=CC(F)=CC=3)CC2)C(=O)NC1 RIOZXKPJYKSKJV-UHFFFAOYSA-N 0.000 description 1
- 229950002809 fluspiperone Drugs 0.000 description 1
- 229960003532 fluspirilene Drugs 0.000 description 1
- 229950009354 flutazolam Drugs 0.000 description 1
- 229950005861 flutemazepam Drugs 0.000 description 1
- 229950003654 flutiazin Drugs 0.000 description 1
- WMWTYOKRWGGJOA-CENSZEJFSA-N fluticasone propionate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(OC(=O)CC)[C@@]2(C)C[C@@H]1O WMWTYOKRWGGJOA-CENSZEJFSA-N 0.000 description 1
- 229960000289 fluticasone propionate Drugs 0.000 description 1
- 229950009348 flutizenol Drugs 0.000 description 1
- 229950009917 flutonidine Drugs 0.000 description 1
- 229950009299 flutoprazepam Drugs 0.000 description 1
- 229950004565 flutroline Drugs 0.000 description 1
- 229950008319 flutropium bromide Drugs 0.000 description 1
- FNUZASGZEHGWQM-RJRMRWARSA-M flutropium bromide Chemical compound C[N@+](CCF)([C@H](CC1)C2)[C@@H]1C[C@H]2OC(C(C1=CC=CC=C1)(C1=CC=CC=C1)O)=O.[Br-] FNUZASGZEHGWQM-RJRMRWARSA-M 0.000 description 1
- CJOFXWAVKWHTFT-XSFVSMFZSA-N fluvoxamine Chemical compound COCCCC\C(=N/OCCN)C1=CC=C(C(F)(F)F)C=C1 CJOFXWAVKWHTFT-XSFVSMFZSA-N 0.000 description 1
- 229960004038 fluvoxamine Drugs 0.000 description 1
- 229950010709 fluzinamide Drugs 0.000 description 1
- 229950010017 fluzoperine Drugs 0.000 description 1
- 102000006815 folate receptor Human genes 0.000 description 1
- 108020005243 folate receptor Proteins 0.000 description 1
- FZRNEERXGKQYPH-UHFFFAOYSA-N folescutol Chemical compound C1=2C=C(O)C(O)=CC=2OC(=O)C=C1CN1CCOCC1 FZRNEERXGKQYPH-UHFFFAOYSA-N 0.000 description 1
- 229950006891 folescutol Drugs 0.000 description 1
- 229950005632 fomidacillin Drugs 0.000 description 1
- KSNNEUZOAFRTDS-UHFFFAOYSA-N fominoben Chemical compound ClC=1C=CC=C(NC(=O)C=2C=CC=CC=2)C=1CN(C)CC(=O)N1CCOCC1 KSNNEUZOAFRTDS-UHFFFAOYSA-N 0.000 description 1
- 229960004594 fominoben Drugs 0.000 description 1
- CVHGCWVMTZWGAY-UHFFFAOYSA-N fomocaine Chemical compound C=1C=C(COC=2C=CC=CC=2)C=CC=1CCCN1CCOCC1 CVHGCWVMTZWGAY-UHFFFAOYSA-N 0.000 description 1
- 229950003051 fomocaine Drugs 0.000 description 1
- 229950001822 fopirtoline Drugs 0.000 description 1
- 229950004217 forfenimex Drugs 0.000 description 1
- AMVODTGMYSRMNP-GNIMZFFESA-N formebolone Chemical compound C1CC2=CC(=O)C(C=O)=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@H]2O AMVODTGMYSRMNP-GNIMZFFESA-N 0.000 description 1
- 229950010292 formebolone Drugs 0.000 description 1
- SGSYPSYCGPLSML-UHFFFAOYSA-N formetorex Chemical compound O=CNC(C)CC1=CC=CC=C1 SGSYPSYCGPLSML-UHFFFAOYSA-N 0.000 description 1
- 229950008855 formetorex Drugs 0.000 description 1
- 229960000671 formocortal Drugs 0.000 description 1
- QNXUUBBKHBYRFW-QWAPGEGQSA-N formocortal Chemical compound C1C(C=O)=C2C=C(OCCCl)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)COC(=O)C)[C@@]1(C)C[C@@H]2O QNXUUBBKHBYRFW-QWAPGEGQSA-N 0.000 description 1
- 229960002848 formoterol Drugs 0.000 description 1
- BPZSYCZIITTYBL-UHFFFAOYSA-N formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-UHFFFAOYSA-N 0.000 description 1
- 229950010605 fosarilate Drugs 0.000 description 1
- 229950006306 fosazepam Drugs 0.000 description 1
- JMYCGCXYZZHWMO-UHFFFAOYSA-N fosazepam Chemical compound N=1CC(=O)N(CP(C)(=O)C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 JMYCGCXYZZHWMO-UHFFFAOYSA-N 0.000 description 1
- 229960005102 foscarnet Drugs 0.000 description 1
- 229960001837 foscolic acid Drugs 0.000 description 1
- 229950009025 fosenazide Drugs 0.000 description 1
- 229960000297 fosfestrol Drugs 0.000 description 1
- 229960000308 fosfomycin Drugs 0.000 description 1
- YMDXZJFXQJVXBF-STHAYSLISA-N fosfomycin Chemical compound C[C@@H]1O[C@@H]1P(O)(O)=O YMDXZJFXQJVXBF-STHAYSLISA-N 0.000 description 1
- 229940112424 fosfonet Drugs 0.000 description 1
- 229950010892 fosfosal Drugs 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 229950000130 fosmenic acid Drugs 0.000 description 1
- 229950006501 fosmidomycin Drugs 0.000 description 1
- GJXWDTUCERCKIX-UHFFFAOYSA-N fosmidomycin Chemical compound O=CN(O)CCCP(O)(O)=O GJXWDTUCERCKIX-UHFFFAOYSA-N 0.000 description 1
- FVYRUSCZCWSFLT-UHFFFAOYSA-N fostedil Chemical compound C1=CC(CP(=O)(OCC)OCC)=CC=C1C1=NC2=CC=CC=C2S1 FVYRUSCZCWSFLT-UHFFFAOYSA-N 0.000 description 1
- 229950006562 fostedil Drugs 0.000 description 1
- 229950010404 fostriecin Drugs 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 229950002403 frabuprofen Drugs 0.000 description 1
- 229950008364 frentizole Drugs 0.000 description 1
- 229950007985 fronepidil Drugs 0.000 description 1
- 229950004738 froxiprost Drugs 0.000 description 1
- 229950000220 ftaxilide Drugs 0.000 description 1
- 229950006548 ftivazide Drugs 0.000 description 1
- 229950009715 ftormetazine Drugs 0.000 description 1
- 229950004824 ftorpropazine Drugs 0.000 description 1
- 229950008052 fubrogonium iodide Drugs 0.000 description 1
- 229950004787 fuprazole Drugs 0.000 description 1
- DLTWLXUYSLIIQN-UHFFFAOYSA-N furacrinic acid Chemical compound C1=C(C)C(C(=O)C(=C)CC)=CC2=C1OC(C(O)=O)=C2 DLTWLXUYSLIIQN-UHFFFAOYSA-N 0.000 description 1
- 229950004052 furacrinic acid Drugs 0.000 description 1
- 229950004998 furafylline Drugs 0.000 description 1
- RWOLIGKRDWLZSV-OWOJBTEDSA-N furalazine Chemical compound N1=NC(N)=NC=C1\C=C\C1=CC=C([N+]([O-])=O)O1 RWOLIGKRDWLZSV-OWOJBTEDSA-N 0.000 description 1
- 229950007048 furalazine Drugs 0.000 description 1
- 229950000337 furaltadone Drugs 0.000 description 1
- YKASHPSKFYVZRC-UHFFFAOYSA-M furan-2-ylmethyl(trimethyl)azanium;iodide Chemical compound [I-].C[N+](C)(C)CC1=CC=CO1 YKASHPSKFYVZRC-UHFFFAOYSA-M 0.000 description 1
- 229950008156 furaprofen Drugs 0.000 description 1
- 229950010710 furazabol Drugs 0.000 description 1
- 229960001625 furazolidone Drugs 0.000 description 1
- PLHJDBGFXBMTGZ-WEVVVXLNSA-N furazolidone Chemical compound O1C([N+](=O)[O-])=CC=C1\C=N\N1C(=O)OCC1 PLHJDBGFXBMTGZ-WEVVVXLNSA-N 0.000 description 1
- 229950008849 furazolium chloride Drugs 0.000 description 1
- 229950001615 furbucillin Drugs 0.000 description 1
- 229950010951 furcloprofen Drugs 0.000 description 1
- 229950002068 furegrelate Drugs 0.000 description 1
- NNCOZXNZFLUYGG-UHFFFAOYSA-N furethidine Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCOCC1CCCO1 NNCOZXNZFLUYGG-UHFFFAOYSA-N 0.000 description 1
- 229950011066 furethidine Drugs 0.000 description 1
- DLGIIZAHQPTVCJ-UHFFFAOYSA-N furfenorex Chemical compound C=1C=COC=1CN(C)C(C)CC1=CC=CC=C1 DLGIIZAHQPTVCJ-UHFFFAOYSA-N 0.000 description 1
- 229950005457 furfenorex Drugs 0.000 description 1
- 229950008976 furidarone Drugs 0.000 description 1
- 229950004090 furmethoxadone Drugs 0.000 description 1
- 229950006099 furobufen Drugs 0.000 description 1
- 229950005802 furodazole Drugs 0.000 description 1
- 229950010931 furofenac Drugs 0.000 description 1
- 229950006484 furomazine Drugs 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 229950004559 furostilbestrol Drugs 0.000 description 1
- 229950002272 fursalan Drugs 0.000 description 1
- 229950001523 furterene Drugs 0.000 description 1
- 229950008907 furtrethonium iodide Drugs 0.000 description 1
- 229960004675 fusidic acid Drugs 0.000 description 1
- IECPWNUMDGFDKC-MZJAQBGESA-N fusidic acid Chemical compound O[C@@H]([C@@H]12)C[C@H]3\C(=C(/CCC=C(C)C)C(O)=O)[C@@H](OC(C)=O)C[C@]3(C)[C@@]2(C)CC[C@@H]2[C@]1(C)CC[C@@H](O)[C@H]2C IECPWNUMDGFDKC-MZJAQBGESA-N 0.000 description 1
- 229950008744 fuzlocillin Drugs 0.000 description 1
- 229960002870 gabapentin Drugs 0.000 description 1
- 229950000501 gabexate Drugs 0.000 description 1
- 229950004346 gaboxadol Drugs 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 108010042430 galactose receptor Proteins 0.000 description 1
- 229960003980 galantamine Drugs 0.000 description 1
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 description 1
- 229960003054 gallamine Drugs 0.000 description 1
- ICLWTJIMXVISSR-UHFFFAOYSA-N gallamine Chemical compound CCN(CC)CCOC1=CC=CC(OCCN(CC)CC)=C1OCCN(CC)CC ICLWTJIMXVISSR-UHFFFAOYSA-N 0.000 description 1
- 229960005271 gallamine triethiodide Drugs 0.000 description 1
- 229960000457 gallopamil Drugs 0.000 description 1
- 229950001564 galosemide Drugs 0.000 description 1
- 229950011389 galtifenin Drugs 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- VZCCETWTMQHEPK-QNEBEIHSSA-N gamma-linolenic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/CCCCC(O)=O VZCCETWTMQHEPK-QNEBEIHSSA-N 0.000 description 1
- 235000020664 gamma-linolenic acid Nutrition 0.000 description 1
- 229960002733 gamolenic acid Drugs 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 1
- 229950008114 ganglefene Drugs 0.000 description 1
- AUQQZPGNRKTPSQ-UHFFFAOYSA-N gapicomine Chemical compound C=1C=NC=CC=1CNCC1=CC=NC=C1 AUQQZPGNRKTPSQ-UHFFFAOYSA-N 0.000 description 1
- 229950002885 gapicomine Drugs 0.000 description 1
- 229950010105 gapromidine Drugs 0.000 description 1
- 210000005095 gastrointestinal system Anatomy 0.000 description 1
- 229960003779 gefarnate Drugs 0.000 description 1
- 229950003686 gemazocine Drugs 0.000 description 1
- 229950002725 gemcadiol Drugs 0.000 description 1
- 229960003480 gemeprost Drugs 0.000 description 1
- KYBOHGVERHWSSV-VNIVIJDLSA-N gemeprost Chemical compound CCCCC(C)(C)[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCC\C=C\C(=O)OC KYBOHGVERHWSSV-VNIVIJDLSA-N 0.000 description 1
- 229960003627 gemfibrozil Drugs 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- RHRAMPXHWHSKQB-UHFFFAOYSA-N gentamicin B Natural products O1CC(O)(C)C(NC)C(O)C1OC1C(O)C(OC2C(C(O)C(O)C(CN)O2)O)C(N)CC1N RHRAMPXHWHSKQB-UHFFFAOYSA-N 0.000 description 1
- 229960001235 gentian violet Drugs 0.000 description 1
- 229960005059 gepefrine Drugs 0.000 description 1
- WTDGMHYYGNJEKQ-ZETCQYMHSA-N gepefrine Chemical compound C[C@H](N)CC1=CC=CC(O)=C1 WTDGMHYYGNJEKQ-ZETCQYMHSA-N 0.000 description 1
- 229960000647 gepirone Drugs 0.000 description 1
- QOIGKGMMAGJZNZ-UHFFFAOYSA-N gepirone Chemical compound O=C1CC(C)(C)CC(=O)N1CCCCN1CCN(C=2N=CC=CN=2)CC1 QOIGKGMMAGJZNZ-UHFFFAOYSA-N 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- 229960000346 gliclazide Drugs 0.000 description 1
- 229950001143 glicondamide Drugs 0.000 description 1
- 229950010476 glidazamide Drugs 0.000 description 1
- 229950002106 gliflumide Drugs 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- 229960003468 gliquidone Drugs 0.000 description 1
- 229950009871 glisamuride Drugs 0.000 description 1
- NSJYMFYVNWVGON-UHFFFAOYSA-N glisentide Chemical compound COC1=CC=CC=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCC2)C=C1 NSJYMFYVNWVGON-UHFFFAOYSA-N 0.000 description 1
- 229950008402 glisentide Drugs 0.000 description 1
- 229950010747 glisindamide Drugs 0.000 description 1
- GZKDXUIWCNCNBJ-UHFFFAOYSA-N glisolamide Chemical compound O1C(C)=CC(C(=O)NCCC=2C=CC(=CC=2)S(=O)(=O)NC(=O)NC2CCCCC2)=N1 GZKDXUIWCNCNBJ-UHFFFAOYSA-N 0.000 description 1
- 229950005319 glisolamide Drugs 0.000 description 1
- ZKUDBRCEOBOWLF-UHFFFAOYSA-N glisoxepide Chemical compound O1C(C)=CC(C(=O)NCCC=2C=CC(=CC=2)S(=O)(=O)NC(=O)NN2CCCCCC2)=N1 ZKUDBRCEOBOWLF-UHFFFAOYSA-N 0.000 description 1
- 229960003236 glisoxepide Drugs 0.000 description 1
- 229950007336 gloxazone Drugs 0.000 description 1
- 229950000189 gloximonam Drugs 0.000 description 1
- LGAJOMLFGCSBFF-XVBLYABRSA-N glucametacin Chemical compound COC1=CC2=C(C=C1)N(C(=O)C1=CC=C(Cl)C=C1)C(C)=C2CC(=O)N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O LGAJOMLFGCSBFF-XVBLYABRSA-N 0.000 description 1
- 229960004410 glucametacin Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 229950006371 glucosulfamide Drugs 0.000 description 1
- SQQCWHCJRWYRLB-AGNGBHFPSA-N glucosulfone Chemical compound C1=CC(NC([C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO)S(O)(=O)=O)=CC=C1S(=O)(=O)C1=CC=C(NC([C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO)S(O)(=O)=O)C=C1 SQQCWHCJRWYRLB-AGNGBHFPSA-N 0.000 description 1
- 229950009858 glucosulfone Drugs 0.000 description 1
- 229950002441 glucurolactone Drugs 0.000 description 1
- 229950004542 glucuronamide Drugs 0.000 description 1
- 229950006554 glunicate Drugs 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 229960000587 glutaral Drugs 0.000 description 1
- WGXUDTHMEITUBO-YFKPBYRVSA-N glutaurine Chemical compound OC(=O)[C@@H](N)CCC(=O)NCCS(O)(=O)=O WGXUDTHMEITUBO-YFKPBYRVSA-N 0.000 description 1
- 229950001776 glutaurine Drugs 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- DVQVBLBKEXITIK-UHFFFAOYSA-N glybuthiazol Chemical compound S1C(C(C)(C)C)=NN=C1NS(=O)(=O)C1=CC=C(N)C=C1 DVQVBLBKEXITIK-UHFFFAOYSA-N 0.000 description 1
- 229950011569 glybuthiazol Drugs 0.000 description 1
- 229950005232 glybuzole Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 229960003711 glyceryl trinitrate Drugs 0.000 description 1
- VOZRXNHHFUQHIL-UHFFFAOYSA-N glycidyl methacrylate Chemical compound CC(=C)C(=O)OCC1CO1 VOZRXNHHFUQHIL-UHFFFAOYSA-N 0.000 description 1
- 229950002888 glyclopyramide Drugs 0.000 description 1
- 125000003827 glycol group Chemical group 0.000 description 1
- 229940015042 glycopyrrolate Drugs 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 150000002338 glycosides Chemical class 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- RIGBPMDIGYBTBJ-UHFFFAOYSA-N glycyclamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 RIGBPMDIGYBTBJ-UHFFFAOYSA-N 0.000 description 1
- 229950005514 glycyclamide Drugs 0.000 description 1
- NFRPNQDSKJJQGV-UHFFFAOYSA-N glyhexamide Chemical compound C=1C=C2CCCC2=CC=1S(=O)(=O)NC(=O)NC1CCCCC1 NFRPNQDSKJJQGV-UHFFFAOYSA-N 0.000 description 1
- 229950008290 glyhexamide Drugs 0.000 description 1
- QFWPJPIVLCBXFJ-UHFFFAOYSA-N glymidine Chemical compound N1=CC(OCCOC)=CN=C1NS(=O)(=O)C1=CC=CC=C1 QFWPJPIVLCBXFJ-UHFFFAOYSA-N 0.000 description 1
- 229960004440 glymidine Drugs 0.000 description 1
- 229950007246 glyoctamide Drugs 0.000 description 1
- RHQSNARBXHRBNP-UHFFFAOYSA-N glypinamide Chemical compound C1=CC(Cl)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 RHQSNARBXHRBNP-UHFFFAOYSA-N 0.000 description 1
- 229950009188 glypinamide Drugs 0.000 description 1
- 229950001670 glyprothiazol Drugs 0.000 description 1
- 229950003831 glysobuzole Drugs 0.000 description 1
- 229940083577 gold sodium thiosulfate Drugs 0.000 description 1
- MFWNKCLOYSRHCJ-BTTYYORXSA-N granisetron Chemical compound C1=CC=C2C(C(=O)N[C@H]3C[C@H]4CCC[C@@H](C3)N4C)=NN(C)C2=C1 MFWNKCLOYSRHCJ-BTTYYORXSA-N 0.000 description 1
- 229960003727 granisetron Drugs 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- WTDYJDLUNYALPM-UHFFFAOYSA-N guabenxan Chemical compound O1CCOC2=CC(CN=C(N)N)=CC=C21 WTDYJDLUNYALPM-UHFFFAOYSA-N 0.000 description 1
- 229950005799 guabenxan Drugs 0.000 description 1
- HSJFYRYGGKLQBT-UHFFFAOYSA-N guacetisal Chemical compound COC1=CC=CC=C1OC(=O)C1=CC=CC=C1OC(C)=O HSJFYRYGGKLQBT-UHFFFAOYSA-N 0.000 description 1
- 229960004478 guacetisal Drugs 0.000 description 1
- 229950000937 guafecainol Drugs 0.000 description 1
- 229950000342 guaiactamine Drugs 0.000 description 1
- PPVFOZYARYOARE-UHFFFAOYSA-N guaiapate Chemical compound COC1=CC=CC=C1OCCOCCOCCN1CCCCC1 PPVFOZYARYOARE-UHFFFAOYSA-N 0.000 description 1
- 229950010056 guaiapate Drugs 0.000 description 1
- 229960004808 guaietolin Drugs 0.000 description 1
- 229960002146 guaifenesin Drugs 0.000 description 1
- PSVDIHULUCLEJE-UHFFFAOYSA-N guaimesal Chemical compound COC1=CC=CC=C1OC1(C)OC2=CC=CC=C2C(=O)O1 PSVDIHULUCLEJE-UHFFFAOYSA-N 0.000 description 1
- 229950006160 guaimesal Drugs 0.000 description 1
- 229950002628 guaisteine Drugs 0.000 description 1
- 229950007488 guamecycline Drugs 0.000 description 1
- 229960004553 guanabenz Drugs 0.000 description 1
- WQVAYGCXSJMPRT-UHFFFAOYSA-N guanacline Chemical compound CC1=CCN(CCN=C(N)N)CC1 WQVAYGCXSJMPRT-UHFFFAOYSA-N 0.000 description 1
- 229950006795 guanacline Drugs 0.000 description 1
- HPBNRIOWIXYZFK-UHFFFAOYSA-N guanadrel Chemical compound O1C(CNC(=N)N)COC11CCCCC1 HPBNRIOWIXYZFK-UHFFFAOYSA-N 0.000 description 1
- 229960003845 guanadrel Drugs 0.000 description 1
- ZCVAIGPGEINFCX-UHFFFAOYSA-N guanazodine Chemical compound NC(=N)NCC1CCCCCCN1 ZCVAIGPGEINFCX-UHFFFAOYSA-N 0.000 description 1
- 229960004614 guanazodine Drugs 0.000 description 1
- PKWIYNIDEDLDCJ-UHFFFAOYSA-N guanazole Chemical compound NC1=NNC(N)=N1 PKWIYNIDEDLDCJ-UHFFFAOYSA-N 0.000 description 1
- VZVGEDRCVUKSEL-UHFFFAOYSA-N guancidine Chemical compound CCC(C)(C)N=C(N)NC#N VZVGEDRCVUKSEL-UHFFFAOYSA-N 0.000 description 1
- 229950007639 guancidine Drugs 0.000 description 1
- 229950011434 guanclofine Drugs 0.000 description 1
- 229960003602 guanethidine Drugs 0.000 description 1
- ACGDKVXYNVEAGU-UHFFFAOYSA-N guanethidine Chemical compound NC(N)=NCCN1CCCCCCC1 ACGDKVXYNVEAGU-UHFFFAOYSA-N 0.000 description 1
- 229960002048 guanfacine Drugs 0.000 description 1
- XIHXRRMCNSMUET-UHFFFAOYSA-N guanoclor Chemical compound NC(=N)NNCCOC1=C(Cl)C=CC=C1Cl XIHXRRMCNSMUET-UHFFFAOYSA-N 0.000 description 1
- 229960002201 guanoclor Drugs 0.000 description 1
- 229950000289 guanoctine Drugs 0.000 description 1
- 229960001016 guanoxabenz Drugs 0.000 description 1
- QKIQJNNDIWGVEH-UUILKARUSA-N guanoxabenz Chemical compound ONC(/N)=N/N=C/C1=C(Cl)C=CC=C1Cl QKIQJNNDIWGVEH-UUILKARUSA-N 0.000 description 1
- 229960000760 guanoxan Drugs 0.000 description 1
- HIUVKVDQFXDZHU-UHFFFAOYSA-N guanoxan Chemical compound C1=CC=C2OC(CNC(=N)N)COC2=C1 HIUVKVDQFXDZHU-UHFFFAOYSA-N 0.000 description 1
- 229950010491 guanoxyfen Drugs 0.000 description 1
- 235000019382 gum benzoic Nutrition 0.000 description 1
- 229960002706 gusperimus Drugs 0.000 description 1
- 229960002158 halazepam Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- ZFSXZJXLKAJIGS-UHFFFAOYSA-N halocarban Chemical compound C1=C(Cl)C(C(F)(F)F)=CC(NC(=O)NC=2C=CC(Cl)=CC=2)=C1 ZFSXZJXLKAJIGS-UHFFFAOYSA-N 0.000 description 1
- 229950006625 halocarban Drugs 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 239000002554 heparinoid Substances 0.000 description 1
- 229940025770 heparinoids Drugs 0.000 description 1
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 1
- VUYDGVRIQRPHFX-UHFFFAOYSA-N hesperidin Natural products COc1cc(ccc1O)C2CC(=O)c3c(O)cc(OC4OC(COC5OC(O)C(O)C(O)C5O)C(O)C(O)C4O)cc3O2 VUYDGVRIQRPHFX-UHFFFAOYSA-N 0.000 description 1
- 229960003884 hetacillin Drugs 0.000 description 1
- DXVUYOAEDJXBPY-NFFDBFGFSA-N hetacillin Chemical compound C1([C@@H]2C(=O)N(C(N2)(C)C)[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 DXVUYOAEDJXBPY-NFFDBFGFSA-N 0.000 description 1
- LGIURICQUGJVAQ-UHFFFAOYSA-M hexadecyl-(2-hydroxycyclohexyl)-dimethylazanium;chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCC[N+](C)(C)C1CCCCC1O LGIURICQUGJVAQ-UHFFFAOYSA-M 0.000 description 1
- 229940084108 hexafluorenium bromide Drugs 0.000 description 1
- WBCLXFIDEDJGCC-UHFFFAOYSA-N hexafluoro-2-butyne Chemical compound FC(F)(F)C#CC(F)(F)F WBCLXFIDEDJGCC-UHFFFAOYSA-N 0.000 description 1
- VBZWSGALLODQNC-UHFFFAOYSA-N hexafluoroacetone Chemical compound FC(F)(F)C(=O)C(F)(F)F VBZWSGALLODQNC-UHFFFAOYSA-N 0.000 description 1
- WMIYKQLTONQJES-UHFFFAOYSA-N hexafluoroethane Chemical compound FC(F)(F)C(F)(F)F WMIYKQLTONQJES-UHFFFAOYSA-N 0.000 description 1
- HCDGVLDPFQMKDK-UHFFFAOYSA-N hexafluoropropylene Chemical compound FC(F)=C(F)C(F)(F)F HCDGVLDPFQMKDK-UHFFFAOYSA-N 0.000 description 1
- WDEFPRUEZRUYNW-UHFFFAOYSA-L hexafluronium bromide Chemical compound [Br-].[Br-].C12=CC=CC=C2C2=CC=CC=C2C1[N+](C)(C)CCCCCC[N+](C)(C)C1C2=CC=CC=C2C2=CC=CC=C21 WDEFPRUEZRUYNW-UHFFFAOYSA-L 0.000 description 1
- YECBTZNXNRHKJD-UHFFFAOYSA-E hexalithium;antimony(3+);2-sulfidobutanedioate Chemical compound [Li+].[Li+].[Li+].[Li+].[Li+].[Li+].[O-]C(=O)CC(C([O-])=O)S[Sb](SC(CC([O-])=O)C([O-])=O)SC(CC([O-])=O)C([O-])=O YECBTZNXNRHKJD-UHFFFAOYSA-E 0.000 description 1
- 150000002402 hexoses Chemical class 0.000 description 1
- 229940006607 hirudin Drugs 0.000 description 1
- WQPDUTSPKFMPDP-OUMQNGNKSA-N hirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(OS(O)(=O)=O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N2)=O)CSSC1)C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)CSSC1)C(C)C)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 WQPDUTSPKFMPDP-OUMQNGNKSA-N 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 229940099552 hyaluronan Drugs 0.000 description 1
- KIUKXJAPPMFGSW-MNSSHETKSA-N hyaluronan Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)C1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H](C(O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-MNSSHETKSA-N 0.000 description 1
- GPGMRSSBVJNWRA-UHFFFAOYSA-N hydrochloride hydrofluoride Chemical compound F.Cl GPGMRSSBVJNWRA-UHFFFAOYSA-N 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- FMPJXUZSXKJUQI-UHFFFAOYSA-N hydron;3-(5-nitrofuran-2-yl)-5,6-dihydroimidazo[2,1-b][1,3]thiazole;chloride Chemical compound Cl.O1C([N+](=O)[O-])=CC=C1C1=CSC2=NCCN12 FMPJXUZSXKJUQI-UHFFFAOYSA-N 0.000 description 1
- PJBQYZZKGNOKNJ-UHFFFAOYSA-M hydron;5-[(2-methylpyridin-1-ium-1-yl)methyl]-2-propylpyrimidin-4-amine;dichloride Chemical compound Cl.[Cl-].NC1=NC(CCC)=NC=C1C[N+]1=CC=CC=C1C PJBQYZZKGNOKNJ-UHFFFAOYSA-M 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 229940050526 hydroxyethylstarch Drugs 0.000 description 1
- CSFWPUWCSPOLJW-UHFFFAOYSA-N hydroxynaphthoquinone Natural products C1=CC=C2C(=O)C(O)=CC(=O)C2=C1 CSFWPUWCSPOLJW-UHFFFAOYSA-N 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 239000012216 imaging agent Substances 0.000 description 1
- 150000002463 imidates Chemical class 0.000 description 1
- JVKYTWZLUFMIKS-UHFFFAOYSA-N imidazo[1,2-a]quinoxaline-2-carboxylic acid Chemical compound C1=CC=C2N3C=C(C(=O)O)N=C3C=NC2=C1 JVKYTWZLUFMIKS-UHFFFAOYSA-N 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 239000000367 immunologic factor Substances 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000001524 infective effect Effects 0.000 description 1
- 230000008798 inflammatory stress Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 229920000592 inorganic polymer Polymers 0.000 description 1
- 230000005732 intercellular adhesion Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 229940117681 interleukin-12 Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229940029339 inulin Drugs 0.000 description 1
- JYJIGFIDKWBXDU-MNNPPOADSA-N inulin Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)OC[C@]1(OC[C@]2(OC[C@]3(OC[C@]4(OC[C@]5(OC[C@]6(OC[C@]7(OC[C@]8(OC[C@]9(OC[C@]%10(OC[C@]%11(OC[C@]%12(OC[C@]%13(OC[C@]%14(OC[C@]%15(OC[C@]%16(OC[C@]%17(OC[C@]%18(OC[C@]%19(OC[C@]%20(OC[C@]%21(OC[C@]%22(OC[C@]%23(OC[C@]%24(OC[C@]%25(OC[C@]%26(OC[C@]%27(OC[C@]%28(OC[C@]%29(OC[C@]%30(OC[C@]%31(OC[C@]%32(OC[C@]%33(OC[C@]%34(OC[C@]%35(OC[C@]%36(O[C@@H]%37[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O%37)O)[C@H]([C@H](O)[C@@H](CO)O%36)O)[C@H]([C@H](O)[C@@H](CO)O%35)O)[C@H]([C@H](O)[C@@H](CO)O%34)O)[C@H]([C@H](O)[C@@H](CO)O%33)O)[C@H]([C@H](O)[C@@H](CO)O%32)O)[C@H]([C@H](O)[C@@H](CO)O%31)O)[C@H]([C@H](O)[C@@H](CO)O%30)O)[C@H]([C@H](O)[C@@H](CO)O%29)O)[C@H]([C@H](O)[C@@H](CO)O%28)O)[C@H]([C@H](O)[C@@H](CO)O%27)O)[C@H]([C@H](O)[C@@H](CO)O%26)O)[C@H]([C@H](O)[C@@H](CO)O%25)O)[C@H]([C@H](O)[C@@H](CO)O%24)O)[C@H]([C@H](O)[C@@H](CO)O%23)O)[C@H]([C@H](O)[C@@H](CO)O%22)O)[C@H]([C@H](O)[C@@H](CO)O%21)O)[C@H]([C@H](O)[C@@H](CO)O%20)O)[C@H]([C@H](O)[C@@H](CO)O%19)O)[C@H]([C@H](O)[C@@H](CO)O%18)O)[C@H]([C@H](O)[C@@H](CO)O%17)O)[C@H]([C@H](O)[C@@H](CO)O%16)O)[C@H]([C@H](O)[C@@H](CO)O%15)O)[C@H]([C@H](O)[C@@H](CO)O%14)O)[C@H]([C@H](O)[C@@H](CO)O%13)O)[C@H]([C@H](O)[C@@H](CO)O%12)O)[C@H]([C@H](O)[C@@H](CO)O%11)O)[C@H]([C@H](O)[C@@H](CO)O%10)O)[C@H]([C@H](O)[C@@H](CO)O9)O)[C@H]([C@H](O)[C@@H](CO)O8)O)[C@H]([C@H](O)[C@@H](CO)O7)O)[C@H]([C@H](O)[C@@H](CO)O6)O)[C@H]([C@H](O)[C@@H](CO)O5)O)[C@H]([C@H](O)[C@@H](CO)O4)O)[C@H]([C@H](O)[C@@H](CO)O3)O)[C@H]([C@H](O)[C@@H](CO)O2)O)[C@@H](O)[C@H](O)[C@@H](CO)O1 JYJIGFIDKWBXDU-MNNPPOADSA-N 0.000 description 1
- VBUWHHLIZKOSMS-RIWXPGAOSA-N invicorp Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)C(C)C)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 VBUWHHLIZKOSMS-RIWXPGAOSA-N 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229940029355 iodipamide Drugs 0.000 description 1
- DGIAUNUPXILTJW-VRWDCWMNSA-N iodipamide dimeglumine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.OC(=O)C1=C(I)C=C(I)C(NC(=O)CCCCC(=O)NC=2C(=C(C(O)=O)C(I)=CC=2I)I)=C1I DGIAUNUPXILTJW-VRWDCWMNSA-N 0.000 description 1
- 229960004359 iodixanol Drugs 0.000 description 1
- NBQNWMBBSKPBAY-UHFFFAOYSA-N iodixanol Chemical compound IC=1C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C(I)C=1N(C(=O)C)CC(O)CN(C(C)=O)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I NBQNWMBBSKPBAY-UHFFFAOYSA-N 0.000 description 1
- 229960001025 iohexol Drugs 0.000 description 1
- NTHXOOBQLCIOLC-UHFFFAOYSA-N iohexol Chemical compound OCC(O)CN(C(=O)C)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I NTHXOOBQLCIOLC-UHFFFAOYSA-N 0.000 description 1
- 229960004647 iopamidol Drugs 0.000 description 1
- XQZXYNRDCRIARQ-LURJTMIESA-N iopamidol Chemical compound C[C@H](O)C(=O)NC1=C(I)C(C(=O)NC(CO)CO)=C(I)C(C(=O)NC(CO)CO)=C1I XQZXYNRDCRIARQ-LURJTMIESA-N 0.000 description 1
- 229960000824 iopentol Drugs 0.000 description 1
- IUNJANQVIJDFTQ-UHFFFAOYSA-N iopentol Chemical compound COCC(O)CN(C(C)=O)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I IUNJANQVIJDFTQ-UHFFFAOYSA-N 0.000 description 1
- 229960002603 iopromide Drugs 0.000 description 1
- DGAIEPBNLOQYER-UHFFFAOYSA-N iopromide Chemical compound COCC(=O)NC1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)N(C)CC(O)CO)=C1I DGAIEPBNLOQYER-UHFFFAOYSA-N 0.000 description 1
- 229960000929 iotalamic acid Drugs 0.000 description 1
- 229960001707 ioxaglic acid Drugs 0.000 description 1
- TYYBFXNZMFNZJT-UHFFFAOYSA-N ioxaglic acid Chemical compound CNC(=O)C1=C(I)C(N(C)C(C)=O)=C(I)C(C(=O)NCC(=O)NC=2C(=C(C(=O)NCCO)C(I)=C(C(O)=O)C=2I)I)=C1I TYYBFXNZMFNZJT-UHFFFAOYSA-N 0.000 description 1
- VRIVJOXICYMTAG-IYEMJOQQSA-L iron(ii) gluconate Chemical compound [Fe+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O VRIVJOXICYMTAG-IYEMJOQQSA-L 0.000 description 1
- QRWOVIRDHQJFDB-UHFFFAOYSA-N isobutyl cyanoacrylate Chemical compound CC(C)COC(=O)C(=C)C#N QRWOVIRDHQJFDB-UHFFFAOYSA-N 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 229960003350 isoniazid Drugs 0.000 description 1
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 229960000201 isosorbide dinitrate Drugs 0.000 description 1
- MOYKHGMNXAOIAT-JGWLITMVSA-N isosorbide dinitrate Chemical compound [O-][N+](=O)O[C@H]1CO[C@@H]2[C@H](O[N+](=O)[O-])CO[C@@H]21 MOYKHGMNXAOIAT-JGWLITMVSA-N 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical class C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- SKKLOUVUUNMCJE-FQSMHNGLSA-N kanamycin B Chemical compound N[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SKKLOUVUUNMCJE-FQSMHNGLSA-N 0.000 description 1
- 229930182824 kanamycin B Natural products 0.000 description 1
- SKKLOUVUUNMCJE-UHFFFAOYSA-N kanendomycin Natural products NC1C(O)C(O)C(CN)OC1OC1C(O)C(OC2C(C(N)C(O)C(CO)O2)O)C(N)CC1N SKKLOUVUUNMCJE-UHFFFAOYSA-N 0.000 description 1
- 229960004184 ketamine hydrochloride Drugs 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 229910052743 krypton Inorganic materials 0.000 description 1
- DNNSSWSSYDEUBZ-UHFFFAOYSA-N krypton atom Chemical compound [Kr] DNNSSWSSYDEUBZ-UHFFFAOYSA-N 0.000 description 1
- 210000001865 kupffer cell Anatomy 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 102000003835 leukotriene receptors Human genes 0.000 description 1
- 108090000146 leukotriene receptors Proteins 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- IXHBTMCLRNMKHZ-LBPRGKRZSA-N levobunolol Chemical compound O=C1CCCC2=C1C=CC=C2OC[C@@H](O)CNC(C)(C)C IXHBTMCLRNMKHZ-LBPRGKRZSA-N 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229960005287 lincomycin Drugs 0.000 description 1
- OJMMVQQUTAEWLP-KIDUDLJLSA-N lincomycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@@H](C)O)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 OJMMVQQUTAEWLP-KIDUDLJLSA-N 0.000 description 1
- 108700041430 link Proteins 0.000 description 1
- 235000019421 lipase Nutrition 0.000 description 1
- 150000002634 lipophilic molecules Chemical class 0.000 description 1
- 229960005015 local anesthetics Drugs 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 235000018977 lysine Nutrition 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- FNJVKRQYEQVPLK-UHFFFAOYSA-L magnesium;1-oxido-2-[(1-oxidopyridin-1-ium-2-yl)disulfanyl]pyridin-1-ium;sulfate;trihydrate Chemical compound O.O.O.[Mg+2].[O-]S([O-])(=O)=O.[O-][N+]1=CC=CC=C1SSC1=CC=CC=[N+]1[O-] FNJVKRQYEQVPLK-UHFFFAOYSA-L 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960003464 mefenamic acid Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 229960004296 megestrol acetate Drugs 0.000 description 1
- MIKKOBKEXMRYFQ-WZTVWXICSA-N meglumine amidotrizoate Chemical compound C[NH2+]C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CC(=O)NC1=C(I)C(NC(C)=O)=C(I)C(C([O-])=O)=C1I MIKKOBKEXMRYFQ-WZTVWXICSA-N 0.000 description 1
- 229940053382 meglumine antimonate Drugs 0.000 description 1
- XOGYVDXPYVPAAQ-SESJOKTNSA-M meglumine antimoniate Chemical compound O[Sb](=O)=O.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO XOGYVDXPYVPAAQ-SESJOKTNSA-M 0.000 description 1
- 229940100477 meglumine iodipamide Drugs 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229960002409 mepivacaine Drugs 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- APADFLLAXHIMFU-LGIHQUBZSA-L meso-doxacurium chloride Chemical compound [Cl-].[Cl-].COC1=C(OC)C(OC)=CC(C[C@@H]2[N@@+](CCC3=C2C(=C(OC)C(OC)=C3)OC)(C)CCCOC(=O)CCC(=O)OCCC[N@@+]2(C)[C@@H](C3=C(OC)C(OC)=C(OC)C=C3CC2)CC=2C=C(OC)C(OC)=C(OC)C=2)=C1 APADFLLAXHIMFU-LGIHQUBZSA-L 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000003475 metalloproteinase inhibitor Substances 0.000 description 1
- DJGAAPFSPWAYTJ-UHFFFAOYSA-M metamizole sodium Chemical compound [Na+].O=C1C(N(CS([O-])(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 DJGAAPFSPWAYTJ-UHFFFAOYSA-M 0.000 description 1
- 229960001620 methohexital sodium Drugs 0.000 description 1
- KDXZREBVGAGZHS-UHFFFAOYSA-M methohexital sodium Chemical compound [Na+].CCC#CC(C)C1(CC=C)C(=O)N=C([O-])N(C)C1=O KDXZREBVGAGZHS-UHFFFAOYSA-M 0.000 description 1
- 229940069038 methotrimeprazine hydrochloride Drugs 0.000 description 1
- FQUMWACBNIURCJ-RWZVGCGMSA-N methyl (1r,15s,17r,18r,19s,20s)-7-chloro-18-methoxy-17-(3,4,5-trimethoxybenzoyl)oxy-1,3,11,12,14,15,16,17,18,19,20,21-dodecahydroyohimban-19-carboxylate Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C(C5=CC(Cl)=CC=C5N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 FQUMWACBNIURCJ-RWZVGCGMSA-N 0.000 description 1
- FNAMRDZHKYQEBA-YDQNNXAVSA-N methyl (2e,5z)-7-[(1r,2r,3r,5s)-2-[(e,3r)-4-(3-chlorophenoxy)-3-hydroxybut-1-enyl]-3,5-dihydroxycyclopentyl]hepta-2,5-dienoate Chemical compound COC(=O)\C=C\C\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\[C@@H](O)COC1=CC=CC(Cl)=C1 FNAMRDZHKYQEBA-YDQNNXAVSA-N 0.000 description 1
- REFZGTXXBBUTMJ-YDQNNXAVSA-N methyl (2e,5z)-7-[(1r,2r,3r,5s)-3,5-dihydroxy-2-[(e,3r)-3-hydroxy-4-[3-(trifluoromethyl)phenoxy]but-1-enyl]cyclopentyl]hepta-2,5-dienoate Chemical compound COC(=O)\C=C\C\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\[C@@H](O)COC1=CC=CC(C(F)(F)F)=C1 REFZGTXXBBUTMJ-YDQNNXAVSA-N 0.000 description 1
- QQCOAAFKJZXJFP-GANRVJIKSA-N methyl (Z)-7-[(1S,2S,3S,5R)-3,5-dihydroxy-2-[(E,3R)-3-hydroxy-3-methyloct-1-enyl]cyclopentyl]hept-5-enoate Chemical group CCCCC[C@@](C)(O)\C=C\[C@@H]1[C@@H](O)C[C@@H](O)[C@H]1C\C=C/CCCC(=O)OC QQCOAAFKJZXJFP-GANRVJIKSA-N 0.000 description 1
- CIBHDGPIOICRGX-ZQCHCGQNSA-N methyl (z)-7-[(1r,2r,3r)-3-hydroxy-2-[(e)-4-hydroxy-4-methyloct-1-enyl]-5-oxocyclopentyl]hept-4-enoate Chemical compound CCCCC(C)(O)C\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CC\C=C/CCC(=O)OC CIBHDGPIOICRGX-ZQCHCGQNSA-N 0.000 description 1
- PFRBORHNFYMVOA-UHFFFAOYSA-N methyl 6-chloro-2-methyl-1,1-dioxo-7-sulfamoyl-3,4-dihydro-1$l^{6},2,4-benzothiadiazine-3-carboxylate Chemical compound ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)N(C)C(C(=O)OC)NC2=C1 PFRBORHNFYMVOA-UHFFFAOYSA-N 0.000 description 1
- NMYCTBARPUAEQN-UHFFFAOYSA-N methyl 7-(diethylamino)-4-oxo-6-propyl-1h-quinoline-3-carboxylate Chemical compound N1C=C(C(=O)OC)C(=O)C2=C1C=C(N(CC)CC)C(CCC)=C2 NMYCTBARPUAEQN-UHFFFAOYSA-N 0.000 description 1
- PTOJVMZPWPAXER-VFJVYMGBSA-N methyl 7-[(1r,2r,3r)-3-hydroxy-2-[(e,3r)-3-hydroxy-4-phenoxybut-1-enyl]-5-oxocyclopentyl]hepta-4,5-dienoate Chemical compound O[C@@H]1CC(=O)[C@H](CC=C=CCCC(=O)OC)[C@H]1\C=C\[C@@H](O)COC1=CC=CC=C1 PTOJVMZPWPAXER-VFJVYMGBSA-N 0.000 description 1
- XRISENIKJUKIHD-LHQZMKCDSA-N methyl 7-[(1r,2r,3r)-3-hydroxy-2-[(e,4r)-4-hydroxy-4-(1-propylcyclobutyl)but-1-enyl]-5-oxocyclopentyl]heptanoate Chemical compound CCCC1([C@H](O)C\C=C\[C@@H]2[C@H](C(=O)C[C@H]2O)CCCCCCC(=O)OC)CCC1 XRISENIKJUKIHD-LHQZMKCDSA-N 0.000 description 1
- BYNHBQROLKAEDQ-CNDPCGPLSA-N methyl 7-[(1s,2s,3s,5r)-3,5-dihydroxy-2-[(e,3s)-3-hydroxy-4-phenoxybut-1-enyl]cyclopentyl]hepta-4,5-dienoate Chemical compound COC(=O)CCC=C=CC[C@@H]1[C@H](O)C[C@H](O)[C@H]1\C=C\[C@H](O)COC1=CC=CC=C1 BYNHBQROLKAEDQ-CNDPCGPLSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- LRPJFWDUBNJJKE-UHFFFAOYSA-N methyl n-(6-cyclohexylsulfanyl-1h-benzimidazol-2-yl)carbamate Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1SC1CCCCC1 LRPJFWDUBNJJKE-UHFFFAOYSA-N 0.000 description 1
- BPMVRAQIQQEBLN-OBPBNMOMSA-N methyl n-[(e)-(1-hydroxy-4-oxidoquinoxalin-4-ium-2-ylidene)methyl]iminocarbamate Chemical compound C1=CC=C2N(O)C(=C/N=NC(=O)OC)/C=[N+]([O-])C2=C1 BPMVRAQIQQEBLN-OBPBNMOMSA-N 0.000 description 1
- PEVOWKBJMJVUSV-UHFFFAOYSA-N methyl n-[6-[2-(4-fluorophenyl)-1,3-dioxolan-2-yl]-1h-benzimidazol-2-yl]carbamate Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C1(C=2C=CC(F)=CC=2)OCCO1 PEVOWKBJMJVUSV-UHFFFAOYSA-N 0.000 description 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 description 1
- FAARLWTXUUQFSN-UHFFFAOYSA-N methylellagic acid Natural products O1C(=O)C2=CC(O)=C(O)C3=C2C2=C1C(OC)=C(O)C=C2C(=O)O3 FAARLWTXUUQFSN-UHFFFAOYSA-N 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- UIUXUFNYAYAMOE-UHFFFAOYSA-N methylsilane Chemical compound [SiH3]C UIUXUFNYAYAMOE-UHFFFAOYSA-N 0.000 description 1
- 229960001566 methyltestosterone Drugs 0.000 description 1
- 229960000316 methyprylon Drugs 0.000 description 1
- 229960003085 meticillin Drugs 0.000 description 1
- 229940091062 metocurine iodide Drugs 0.000 description 1
- 229960000554 metrizamide Drugs 0.000 description 1
- 229960004712 metrizoic acid Drugs 0.000 description 1
- GGGDNPWHMNJRFN-UHFFFAOYSA-N metrizoic acid Chemical compound CC(=O)N(C)C1=C(I)C(NC(C)=O)=C(I)C(C(O)=O)=C1I GGGDNPWHMNJRFN-UHFFFAOYSA-N 0.000 description 1
- 229960000282 metronidazole Drugs 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- 229960002509 miconazole Drugs 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- PLYSCVSCYOQVRP-UHFFFAOYSA-N midazolam hydrochloride Chemical compound Cl.C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F PLYSCVSCYOQVRP-UHFFFAOYSA-N 0.000 description 1
- 229960002853 midazolam hydrochloride Drugs 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 229960005181 morphine Drugs 0.000 description 1
- 229940051875 mucins Drugs 0.000 description 1
- 235000010460 mustard Nutrition 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- WMUOPVGLPLDJIX-UHFFFAOYSA-N n'-(1-benzofuran-2-ylmethyl)-n,n-dimethyl-n'-pyridin-2-ylethane-1,2-diamine Chemical compound C=1C2=CC=CC=C2OC=1CN(CCN(C)C)C1=CC=CC=N1 WMUOPVGLPLDJIX-UHFFFAOYSA-N 0.000 description 1
- ZLPKTCYXMOJGBC-UHFFFAOYSA-N n'-acetyl-3-hydroxy-3,3-diphenylpropanehydrazide Chemical compound C=1C=CC=CC=1C(O)(CC(=O)NNC(=O)C)C1=CC=CC=C1 ZLPKTCYXMOJGBC-UHFFFAOYSA-N 0.000 description 1
- FDJAMXYLIRPRIS-UHFFFAOYSA-N n'-acetylpyridine-2-carbohydrazide Chemical group CC(=O)NNC(=O)C1=CC=CC=N1 FDJAMXYLIRPRIS-UHFFFAOYSA-N 0.000 description 1
- ZIUHHBKFKCYYJD-UHFFFAOYSA-N n,n'-methylenebisacrylamide Chemical compound C=CC(=O)NCNC(=O)C=C ZIUHHBKFKCYYJD-UHFFFAOYSA-N 0.000 description 1
- QEYHZXNEIGHKAV-UHFFFAOYSA-N n,n,2,2,3-pentamethylbicyclo[2.2.1]heptan-3-amine Chemical compound C1CC2C(C)(C)C(N(C)C)(C)C1C2 QEYHZXNEIGHKAV-UHFFFAOYSA-N 0.000 description 1
- FGGFIMIICGZCCJ-UHFFFAOYSA-N n,n-dibutyl-4-hexoxynaphthalene-1-carboximidamide Chemical compound C1=CC=C2C(OCCCCCC)=CC=C(C(=N)N(CCCC)CCCC)C2=C1 FGGFIMIICGZCCJ-UHFFFAOYSA-N 0.000 description 1
- QPHACUMLBDXKIF-UHFFFAOYSA-M n,n-diethyl-1-methylpyridin-1-ium-3-carboxamide;4,7,7-trimethyl-3-oxobicyclo[2.2.1]heptane-2-sulfonate Chemical compound CCN(CC)C(=O)C1=CC=C[N+](C)=C1.C1CC2(C)C(=O)C(S([O-])(=O)=O)C1C2(C)C QPHACUMLBDXKIF-UHFFFAOYSA-M 0.000 description 1
- OTEAKJIIJRDXBV-UHFFFAOYSA-N n,n-diethyl-2-(2-methoxyphenoxy)ethanamine Chemical compound CCN(CC)CCOC1=CC=CC=C1OC OTEAKJIIJRDXBV-UHFFFAOYSA-N 0.000 description 1
- QZYFQEXKWXCXJQ-UHFFFAOYSA-N n,n-dimethyl-1-spiro[1,3-dioxolane-2,11'-5,6-dihydrodibenzo[1,2-a:1',2'-e][7]annulene]-4-ylmethanamine Chemical compound O1C(CN(C)C)COC21C1=CC=CC=C1CCC1=CC=CC=C12 QZYFQEXKWXCXJQ-UHFFFAOYSA-N 0.000 description 1
- WZXJEMXDECWINY-UHFFFAOYSA-N n,n-dimethyl-2,3,4,9-tetrahydro-1h-carbazol-3-amine Chemical compound N1C2=CC=CC=C2C2=C1CCC(N(C)C)C2 WZXJEMXDECWINY-UHFFFAOYSA-N 0.000 description 1
- FLIFVPQCMBENHZ-UHFFFAOYSA-N n,n-dimethyl-4-[2-[3-methyl-4-(4-phenylphenyl)-1,3-thiazol-3-ium-2-yl]ethenyl]aniline Chemical compound C1=CC(N(C)C)=CC=C1\C=C\C1=[N+](C)C(C=2C=CC(=CC=2)C=2C=CC=CC=2)=CS1 FLIFVPQCMBENHZ-UHFFFAOYSA-N 0.000 description 1
- ODRQYAHPAJZPQP-UHFFFAOYSA-N n-(2,3-dihydro-1,4-benzodioxin-3-ylmethyl)butan-1-amine Chemical compound C1=CC=C2OC(CNCCCC)COC2=C1 ODRQYAHPAJZPQP-UHFFFAOYSA-N 0.000 description 1
- GDDYCOSWVJRUHM-UHFFFAOYSA-N n-(2,4-dichlorophenyl)-3-piperidin-1-ylbutanamide Chemical compound C1CCCCN1C(C)CC(=O)NC1=CC=C(Cl)C=C1Cl GDDYCOSWVJRUHM-UHFFFAOYSA-N 0.000 description 1
- LZQLXSNYZUENIF-UHFFFAOYSA-N n-(2,6-dichlorophenyl)-4,5-dihydro-1,3-oxazol-2-amine Chemical compound ClC1=CC=CC(Cl)=C1NC1=NCCO1 LZQLXSNYZUENIF-UHFFFAOYSA-N 0.000 description 1
- NVASAXNGHUPVJX-UHFFFAOYSA-N n-(2,6-dimethylphenyl)-1-(2-hydroxyethyl)piperidine-2-carboxamide Chemical compound CC1=CC=CC(C)=C1NC(=O)C1N(CCO)CCCC1 NVASAXNGHUPVJX-UHFFFAOYSA-N 0.000 description 1
- MLUILGIMZAVBDU-UHFFFAOYSA-N n-(2-chlorophenyl)-2-[2-(diethylamino)ethyl-ethylamino]acetamide Chemical compound CCN(CC)CCN(CC)CC(=O)NC1=CC=CC=C1Cl MLUILGIMZAVBDU-UHFFFAOYSA-N 0.000 description 1
- NEXBVZAHMLKKIJ-UHFFFAOYSA-N n-(2-cyanoethyl)-3-methyl-4-oxido-1-oxoquinoxalin-1-ium-2-carboxamide Chemical compound C1=CC=C2N([O-])C(C)=C(C(=O)NCCC#N)[N+](=O)C2=C1 NEXBVZAHMLKKIJ-UHFFFAOYSA-N 0.000 description 1
- WJOQWLQQCYYQBE-UHFFFAOYSA-N n-(2-methylphenyl)-2-pyrrolidin-1-ylpropanamide Chemical compound C=1C=CC=C(C)C=1NC(=O)C(C)N1CCCC1 WJOQWLQQCYYQBE-UHFFFAOYSA-N 0.000 description 1
- JLBDOSAQLCZTSX-UHFFFAOYSA-N n-(3-benzoylphenyl)-1,1-difluoromethanesulfonamide Chemical compound FC(F)S(=O)(=O)NC1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 JLBDOSAQLCZTSX-UHFFFAOYSA-N 0.000 description 1
- UJUHYRDNSIRHHG-UHFFFAOYSA-N n-(4-ethoxyphenyl)-2-hydroxyacetamide Chemical compound CCOC1=CC=C(NC(=O)CO)C=C1 UJUHYRDNSIRHHG-UHFFFAOYSA-N 0.000 description 1
- VDKXSMCTWDWLSB-UHFFFAOYSA-N n-(5-butyl-1,3,4-thiadiazol-2-yl)-4-chlorobenzenesulfonamide Chemical compound S1C(CCCC)=NN=C1NS(=O)(=O)C1=CC=C(Cl)C=C1 VDKXSMCTWDWLSB-UHFFFAOYSA-N 0.000 description 1
- WMJNWYCBXNTPQN-UHFFFAOYSA-N n-(5-cyano-4-oxopyrimidin-1-yl)acetamide Chemical compound CC(=O)NN1C=NC(=O)C(C#N)=C1 WMJNWYCBXNTPQN-UHFFFAOYSA-N 0.000 description 1
- UYIZEUBMWCOJFF-UHFFFAOYSA-N n-(5-fluoro-2-methylphenyl)-4,5-dihydro-1h-imidazol-2-amine Chemical compound CC1=CC=C(F)C=C1NC1=NCCN1 UYIZEUBMWCOJFF-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- PKUBDVAOXLEWBF-GHMZBOCLSA-N n-[(1r,2r)-1-(4-acetylphenyl)-1,3-dihydroxypropan-2-yl]-2,2-dichloroacetamide Chemical compound CC(=O)C1=CC=C([C@@H](O)[C@@H](CO)NC(=O)C(Cl)Cl)C=C1 PKUBDVAOXLEWBF-GHMZBOCLSA-N 0.000 description 1
- CKOLETHYECDWSS-KRWDZBQOSA-N n-[(1s)-1-(5-fluoro-2-methoxyphenyl)ethyl]-2-[4-[[5-(2-methylpropyl)pyrimidin-2-yl]sulfamoyl]phenyl]acetamide Chemical compound COC1=CC=C(F)C=C1[C@H](C)NC(=O)CC1=CC=C(S(=O)(=O)NC=2N=CC(CC(C)C)=CN=2)C=C1 CKOLETHYECDWSS-KRWDZBQOSA-N 0.000 description 1
- IDINUJSAMVOPCM-INIZCTEOSA-N n-[(1s)-2-[4-(3-aminopropylamino)butylamino]-1-hydroxy-2-oxoethyl]-7-(diaminomethylideneamino)heptanamide Chemical compound NCCCNCCCCNC(=O)[C@H](O)NC(=O)CCCCCCN=C(N)N IDINUJSAMVOPCM-INIZCTEOSA-N 0.000 description 1
- OZAREULYFHAGNW-PMACEKPBSA-N n-[(2r,3r)-2,3-diphenylcyclopropyl]-2-pyrrolidin-1-ylacetamide Chemical compound C1([C@H]([C@@H]1C=1C=CC=CC=1)C=1C=CC=CC=1)NC(=O)CN1CCCC1 OZAREULYFHAGNW-PMACEKPBSA-N 0.000 description 1
- LLJDJQYGZBQFIF-DHZHZOJOSA-N n-[(4-amino-2-methylpyrimidin-5-yl)methyl]-n-[(1e)-1-(2-oxo-1,3-oxathian-4-ylidene)ethyl]formamide Chemical compound C/1COC(=O)SC\1=C(/C)N(C=O)CC1=CN=C(C)N=C1N LLJDJQYGZBQFIF-DHZHZOJOSA-N 0.000 description 1
- SRLPZQAEBMZCIJ-UHFFFAOYSA-N n-[(4-chlorophenyl)carbamoyl]-2-(dimethylamino)-6-fluorobenzamide Chemical compound CN(C)C1=CC=CC(F)=C1C(=O)NC(=O)NC1=CC=C(Cl)C=C1 SRLPZQAEBMZCIJ-UHFFFAOYSA-N 0.000 description 1
- WKRAEDUMAWVCOC-UHFFFAOYSA-N n-[(5-ethoxy-2,3-dihydro-1,4-benzodioxin-3-yl)methyl]butan-1-amine Chemical compound C1=CC(OCC)=C2OC(CNCCCC)COC2=C1 WKRAEDUMAWVCOC-UHFFFAOYSA-N 0.000 description 1
- JRRUSQGIRBEMRN-HNNXBMFYSA-N n-[(7s)-3-hydroxy-1,2,10-trimethoxy-9-oxo-6,7-dihydro-5h-benzo[a]heptalen-7-yl]acetamide Chemical compound O=C1C(OC)=CC=C2C3=C(OC)C(OC)=C(O)C=C3CC[C@H](NC(C)=O)C2=C1 JRRUSQGIRBEMRN-HNNXBMFYSA-N 0.000 description 1
- YLWYNCPRXFCNLQ-WYIUXTECSA-N n-[(e)-[(e)-but-2-enylidene]amino]pyridine-4-carboxamide Chemical compound C\C=C\C=N\NC(=O)C1=CC=NC=C1 YLWYNCPRXFCNLQ-WYIUXTECSA-N 0.000 description 1
- NJSMWLQOCQIOPE-OCHFTUDZSA-N n-[(e)-[10-[(e)-(4,5-dihydro-1h-imidazol-2-ylhydrazinylidene)methyl]anthracen-9-yl]methylideneamino]-4,5-dihydro-1h-imidazol-2-amine Chemical compound N1CCN=C1N\N=C\C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1\C=N\NC1=NCCN1 NJSMWLQOCQIOPE-OCHFTUDZSA-N 0.000 description 1
- ZWKFENYDXISLGK-UHFFFAOYSA-N n-[2-(3,4-dihydroxyphenyl)ethyl]adamantane-1-carboxamide Chemical compound C1=C(O)C(O)=CC=C1CCNC(=O)C1(C2)CC(C3)CC2CC3C1 ZWKFENYDXISLGK-UHFFFAOYSA-N 0.000 description 1
- GXLWOFJVIBEMCS-UHFFFAOYSA-N n-[2-(diethylamino)ethyl]-2-(4-phenylphenyl)propanamide Chemical compound C1=CC(C(C)C(=O)NCCN(CC)CC)=CC=C1C1=CC=CC=C1 GXLWOFJVIBEMCS-UHFFFAOYSA-N 0.000 description 1
- NPORGATXHNRFGX-UHFFFAOYSA-N n-[2-(diethylamino)ethyl]-2-prop-2-enoxy-4-(trifluoromethyl)benzamide Chemical compound CCN(CC)CCNC(=O)C1=CC=C(C(F)(F)F)C=C1OCC=C NPORGATXHNRFGX-UHFFFAOYSA-N 0.000 description 1
- OGWJLWKFDHLZHV-UHFFFAOYSA-N n-[2-(diethylamino)ethyl]-4-hydroxy-2-oxochromene-3-carboxamide Chemical compound C1=CC=C2OC(=O)C(C(=O)NCCN(CC)CC)=C(O)C2=C1 OGWJLWKFDHLZHV-UHFFFAOYSA-N 0.000 description 1
- XZHMFCUWVDUYBV-UHFFFAOYSA-N n-[2-[4-(1-benzofuran-7-yl)piperazin-1-yl]ethyl]-n-methyl-4-propan-2-ylbenzamide Chemical compound C1=CC(C(C)C)=CC=C1C(=O)N(C)CCN1CCN(C=2C=3OC=CC=3C=CC=2)CC1 XZHMFCUWVDUYBV-UHFFFAOYSA-N 0.000 description 1
- GBKGGLHRMGPFMB-UHFFFAOYSA-N n-[2-[4-(cyclohexylcarbamoylsulfamoyl)phenyl]ethyl]-3-oxo-1h-isoindole-2-carboxamide Chemical compound C=1C=C(CCNC(=O)N2C(C3=CC=CC=C3C2)=O)C=CC=1S(=O)(=O)NC(=O)NC1CCCCC1 GBKGGLHRMGPFMB-UHFFFAOYSA-N 0.000 description 1
- JSELIWGNAHBPAE-UHFFFAOYSA-N n-[2-[4-[2-(diethylamino)ethoxy]-4-phenylpiperidin-1-yl]ethyl]aniline Chemical compound C1CC(OCCN(CC)CC)(C=2C=CC=CC=2)CCN1CCNC1=CC=CC=C1 JSELIWGNAHBPAE-UHFFFAOYSA-N 0.000 description 1
- JZQYAVBXJCQSOK-UHFFFAOYSA-N n-[3-(diethylamino)-2-hydroxypropyl]-3-methoxy-1-phenylindole-2-carboxamide Chemical compound CCN(CC)CC(O)CNC(=O)C1=C(OC)C2=CC=CC=C2N1C1=CC=CC=C1 JZQYAVBXJCQSOK-UHFFFAOYSA-N 0.000 description 1
- NFZJQAPVHJPJMD-UHFFFAOYSA-N n-[4-(2,2,2-trichloro-1-hydroxyethoxy)phenyl]acetamide Chemical compound CC(=O)NC1=CC=C(OC(O)C(Cl)(Cl)Cl)C=C1 NFZJQAPVHJPJMD-UHFFFAOYSA-N 0.000 description 1
- FXJAOWANXXJWGJ-UHFFFAOYSA-N n-[4-(2-methyl-1h-imidazol-5-yl)phenyl]-n'-propan-2-ylmethanimidamide Chemical compound C1=CC(NC=NC(C)C)=CC=C1C1=CN=C(C)N1 FXJAOWANXXJWGJ-UHFFFAOYSA-N 0.000 description 1
- DCDXHGMCXGHXBM-PMACEKPBSA-N n-[4-[(4as,10br)-8,9-dimethoxy-2-methyl-3,4,4a,10b-tetrahydro-1h-benzo[c][1,6]naphthyridin-6-yl]phenyl]acetamide Chemical compound N([C@H]1CCN(C)C[C@H]1C=1C=C(C(=CC=11)OC)OC)=C1C1=CC=C(NC(C)=O)C=C1 DCDXHGMCXGHXBM-PMACEKPBSA-N 0.000 description 1
- JNEZCZPNQCQCFK-UHFFFAOYSA-N n-[4-[2-[2-(4-acetamidophenoxy)ethoxy]ethoxy]phenyl]acetamide Chemical compound C1=CC(NC(=O)C)=CC=C1OCCOCCOC1=CC=C(NC(C)=O)C=C1 JNEZCZPNQCQCFK-UHFFFAOYSA-N 0.000 description 1
- GNWHIAXZYUDAFX-UHFFFAOYSA-N n-[4-[3-(trifluoromethyl)anilino]pyridin-3-yl]sulfonylpropanamide Chemical compound CCC(=O)NS(=O)(=O)C1=CN=CC=C1NC1=CC=CC(C(F)(F)F)=C1 GNWHIAXZYUDAFX-UHFFFAOYSA-N 0.000 description 1
- CGUIWXDBHIFQJV-UHFFFAOYSA-N n-[4-[3-[2-(3,4-dimethoxyphenyl)ethylamino]-2-hydroxypropoxy]-3-(1,2-oxazol-5-yl)phenyl]butanamide Chemical compound C=1C=NOC=1C1=CC(NC(=O)CCC)=CC=C1OCC(O)CNCCC1=CC=C(OC)C(OC)=C1 CGUIWXDBHIFQJV-UHFFFAOYSA-N 0.000 description 1
- UPBAOYRENQEPJO-UHFFFAOYSA-N n-[5-[[5-[(3-amino-3-iminopropyl)carbamoyl]-1-methylpyrrol-3-yl]carbamoyl]-1-methylpyrrol-3-yl]-4-formamido-1-methylpyrrole-2-carboxamide Chemical compound CN1C=C(NC=O)C=C1C(=O)NC1=CN(C)C(C(=O)NC2=CN(C)C(C(=O)NCCC(N)=N)=C2)=C1 UPBAOYRENQEPJO-UHFFFAOYSA-N 0.000 description 1
- BTYDXWCTJDAEHA-UHFFFAOYSA-N n-[[1-(cyclopropylmethyl)pyrrolidin-2-yl]methyl]-2-methoxy-5-sulfamoylbenzamide Chemical compound COC1=CC=C(S(N)(=O)=O)C=C1C(=O)NCC1N(CC2CC2)CCC1 BTYDXWCTJDAEHA-UHFFFAOYSA-N 0.000 description 1
- DZSKXGDTCMADPR-UHFFFAOYSA-N n-[[1-[(4-fluorophenyl)methyl]pyrrolidin-2-yl]methyl]-2-methoxy-5-sulfamoylbenzamide Chemical compound COC1=CC=C(S(N)(=O)=O)C=C1C(=O)NCC1N(CC=2C=CC(F)=CC=2)CCC1 DZSKXGDTCMADPR-UHFFFAOYSA-N 0.000 description 1
- WYZDCUGWXKHESN-UHFFFAOYSA-N n-benzyl-n-methyl-1-phenylmethanamine Chemical compound C=1C=CC=CC=1CN(C)CC1=CC=CC=C1 WYZDCUGWXKHESN-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- DIOQZVSQGTUSAI-UHFFFAOYSA-N n-butylhexane Natural products CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 1
- RTFADALJKSFJDZ-UHFFFAOYSA-N n-cyclohexyl-2-piperazin-1-ylacetamide Chemical compound C1CCCCC1NC(=O)CN1CCNCC1 RTFADALJKSFJDZ-UHFFFAOYSA-N 0.000 description 1
- CPYSWRYVSINXMC-UHFFFAOYSA-N n-ethyl-1,2-benzothiazol-3-amine Chemical compound C1=CC=C2C(NCC)=NSC2=C1 CPYSWRYVSINXMC-UHFFFAOYSA-N 0.000 description 1
- VXMGLMHPFWGAJO-UHFFFAOYSA-N n-hydroxy-2-(5-methoxy-2-methyl-1h-indol-3-yl)acetamide Chemical compound COC1=CC=C2NC(C)=C(CC(=O)NO)C2=C1 VXMGLMHPFWGAJO-UHFFFAOYSA-N 0.000 description 1
- XVWQQNARVMHZBP-UHFFFAOYSA-N n-methyl-2-phenyl-n-(2-phenylethyl)ethanamine Chemical compound C=1C=CC=CC=1CCN(C)CCC1=CC=CC=C1 XVWQQNARVMHZBP-UHFFFAOYSA-N 0.000 description 1
- YULWJRNIKFFGNU-UHFFFAOYSA-N n-methyl-3-[3-(trifluoromethyl)phenoxy]azetidine-1-carboxamide Chemical compound C1N(C(=O)NC)CC1OC1=CC=CC(C(F)(F)F)=C1 YULWJRNIKFFGNU-UHFFFAOYSA-N 0.000 description 1
- YDSVAKPJAOSZJA-UHFFFAOYSA-N n-methyl-4,5,6,7-tetrahydro-1,3-benzothiazol-6-amine Chemical compound C1C(NC)CCC2=C1SC=N2 YDSVAKPJAOSZJA-UHFFFAOYSA-N 0.000 description 1
- CSDTZUBPSYWZDX-UHFFFAOYSA-N n-pentyl nitrite Chemical compound CCCCCON=O CSDTZUBPSYWZDX-UHFFFAOYSA-N 0.000 description 1
- SGDDXBVRZYARLT-VOTSOKGWSA-N n-propan-2-yl-2-[4-[(e)-3-(3,4,5-trimethoxyphenyl)prop-2-enoyl]piperazin-1-yl]acetamide Chemical compound COC1=C(OC)C(OC)=CC(\C=C\C(=O)N2CCN(CC(=O)NC(C)C)CC2)=C1 SGDDXBVRZYARLT-VOTSOKGWSA-N 0.000 description 1
- GPXLMGHLHQJAGZ-JTDSTZFVSA-N nafcillin Chemical compound C1=CC=CC2=C(C(=O)N[C@@H]3C(N4[C@H](C(C)(C)S[C@@H]43)C(O)=O)=O)C(OCC)=CC=C21 GPXLMGHLHQJAGZ-JTDSTZFVSA-N 0.000 description 1
- 229960000515 nafcillin Drugs 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-M naproxen(1-) Chemical compound C1=C([C@H](C)C([O-])=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-M 0.000 description 1
- 239000004081 narcotic agent Substances 0.000 description 1
- 230000001452 natriuretic effect Effects 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 229960003966 nicotinamide Drugs 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 239000011570 nicotinamide Substances 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 229960000988 nystatin Drugs 0.000 description 1
- VQOXZBDYSJBXMA-NQTDYLQESA-N nystatin A1 Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/CC/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 VQOXZBDYSJBXMA-NQTDYLQESA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- BCCOBQSFUDVTJQ-UHFFFAOYSA-N octafluorocyclobutane Chemical compound FC1(F)C(F)(F)C(F)(F)C1(F)F BCCOBQSFUDVTJQ-UHFFFAOYSA-N 0.000 description 1
- 235000019407 octafluorocyclobutane Nutrition 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229960001027 opium Drugs 0.000 description 1
- SUWZHLCNFQWNPE-LATRNWQMSA-N optochin Chemical compound C([C@H]([C@H](C1)CC)C2)CN1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OCC)C=C21 SUWZHLCNFQWNPE-LATRNWQMSA-N 0.000 description 1
- 125000002092 orthoester group Chemical group 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 150000002924 oxiranes Chemical class 0.000 description 1
- CNANHKGYHBTGAQ-UHFFFAOYSA-N oxolan-2-ylmethyl 2-(2-carbamoylphenoxy)acetate Chemical compound NC(=O)C1=CC=CC=C1OCC(=O)OCC1OCCC1 CNANHKGYHBTGAQ-UHFFFAOYSA-N 0.000 description 1
- CMHHMUWAYWTMGS-UHFFFAOYSA-N oxybuprocaine Chemical compound CCCCOC1=CC(C(=O)OCCN(CC)CC)=CC=C1N CMHHMUWAYWTMGS-UHFFFAOYSA-N 0.000 description 1
- 229960003502 oxybuprocaine Drugs 0.000 description 1
- 230000000242 pagocytic effect Effects 0.000 description 1
- NPIJXCQZLFKBMV-YTGGZNJNSA-L pancuronium bromide Chemical compound [Br-].[Br-].C[N+]1([C@@H]2[C@@H](OC(C)=O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)C)[N+]2(C)CCCCC2)CCCCC1 NPIJXCQZLFKBMV-YTGGZNJNSA-L 0.000 description 1
- 229960003379 pancuronium bromide Drugs 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- RUVINXPYWBROJD-UHFFFAOYSA-N para-methoxyphenyl Natural products COC1=CC=C(C=CC)C=C1 RUVINXPYWBROJD-UHFFFAOYSA-N 0.000 description 1
- 229960003868 paraldehyde Drugs 0.000 description 1
- SQYNKIJPMDEDEG-UHFFFAOYSA-N paraldehyde Chemical compound CC1OC(C)OC(C)O1 SQYNKIJPMDEDEG-UHFFFAOYSA-N 0.000 description 1
- 230000005298 paramagnetic effect Effects 0.000 description 1
- 229960000865 paramethasone acetate Drugs 0.000 description 1
- 230000000849 parathyroid Effects 0.000 description 1
- 239000008414 paregoric Substances 0.000 description 1
- 229940069533 paregoric Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 229960001639 penicillamine Drugs 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 229940056360 penicillin g Drugs 0.000 description 1
- 229960004321 pentaerithrityl tetranitrate Drugs 0.000 description 1
- QIYZKVMAFMDRTP-UHFFFAOYSA-N pentafluoro(trifluoromethyl)-$l^{6}-sulfane Chemical compound FC(F)(F)S(F)(F)(F)(F)F QIYZKVMAFMDRTP-UHFFFAOYSA-N 0.000 description 1
- 229960001412 pentobarbital Drugs 0.000 description 1
- 229940043138 pentosan polysulfate Drugs 0.000 description 1
- 150000002972 pentoses Chemical class 0.000 description 1
- 229960003436 pentoxyverine Drugs 0.000 description 1
- LGUZHRODIJCVOC-UHFFFAOYSA-N perfluoroheptane Chemical class FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F LGUZHRODIJCVOC-UHFFFAOYSA-N 0.000 description 1
- ZJIJAJXFLBMLCK-UHFFFAOYSA-N perfluorohexane Chemical class FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F ZJIJAJXFLBMLCK-UHFFFAOYSA-N 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- XENPFVQKDICWOI-UHFFFAOYSA-N phenazin-10-ium-1-ylmethanediamine;chloride Chemical compound [Cl-].C1=CC=C2N=C3C(C(N)N)=CC=CC3=[NH+]C2=C1 XENPFVQKDICWOI-UHFFFAOYSA-N 0.000 description 1
- 229960005222 phenazone Drugs 0.000 description 1
- 229960003877 pheneturide Drugs 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229960002895 phenylbutazone Drugs 0.000 description 1
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 1
- 229940067605 phosphatidylethanolamines Drugs 0.000 description 1
- 102000023884 phosphatidylserine binding proteins Human genes 0.000 description 1
- 108091008423 phosphatidylserine binding proteins Proteins 0.000 description 1
- 229950007002 phosphocreatine Drugs 0.000 description 1
- XUYJLQHKOGNDPB-UHFFFAOYSA-N phosphonoacetic acid Chemical compound OC(=O)CP(O)(O)=O XUYJLQHKOGNDPB-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- KNCYXPMJDCCGSJ-UHFFFAOYSA-N piperidine-2,6-dione Chemical compound O=C1CCCC(=O)N1 KNCYXPMJDCCGSJ-UHFFFAOYSA-N 0.000 description 1
- 229960002702 piroxicam Drugs 0.000 description 1
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 1
- 201000008814 placenta cancer Diseases 0.000 description 1
- 208000024361 placenta neoplasm Diseases 0.000 description 1
- 229940012957 plasmin Drugs 0.000 description 1
- 102000005162 pleiotrophin Human genes 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 1
- 229920001308 poly(aminoacid) Polymers 0.000 description 1
- 229920002627 poly(phosphazenes) Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 239000010318 polygalacturonic acid Substances 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 1
- 235000013824 polyphenols Nutrition 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 229960002262 profenamine Drugs 0.000 description 1
- CDOZDBSBBXSXLB-UHFFFAOYSA-N profenamine Chemical compound C1=CC=C2N(CC(C)N(CC)CC)C3=CC=CC=C3SC2=C1 CDOZDBSBBXSXLB-UHFFFAOYSA-N 0.000 description 1
- SSOLNOMRVKKSON-UHFFFAOYSA-N proguanil Chemical compound CC(C)\N=C(/N)N=C(N)NC1=CC=C(Cl)C=C1 SSOLNOMRVKKSON-UHFFFAOYSA-N 0.000 description 1
- 229960005385 proguanil Drugs 0.000 description 1
- MKTSXEFLKGUYFZ-KXGFGANYSA-N propan-2-yl (4s,5z)-4-(2,3-dichlorophenyl)-2,6-dimethyl-5-(3h-1,3,4-oxadiazol-2-ylidene)-4h-pyridine-3-carboxylate Chemical compound C1([C@H]2C(/C(C)=NC(C)=C2C(=O)OC(C)C)=C\2OC=NN/2)=CC=CC(Cl)=C1Cl MKTSXEFLKGUYFZ-KXGFGANYSA-N 0.000 description 1
- QZWHWHNCPFEXLL-UHFFFAOYSA-N propan-2-yl n-[2-(1,3-thiazol-4-yl)-3h-benzimidazol-5-yl]carbamate Chemical compound N1C2=CC(NC(=O)OC(C)C)=CC=C2N=C1C1=CSC=N1 QZWHWHNCPFEXLL-UHFFFAOYSA-N 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- MWWATHDPGQKSAR-UHFFFAOYSA-N propyne Chemical compound CC#C MWWATHDPGQKSAR-UHFFFAOYSA-N 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- PXGPLTODNUVGFL-YNNPMVKQSA-N prostaglandin F2alpha Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O PXGPLTODNUVGFL-YNNPMVKQSA-N 0.000 description 1
- PXGPLTODNUVGFL-JZFBHDEDSA-N prostaglandin F2beta Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)C[C@@H](O)[C@@H]1C\C=C/CCCC(O)=O PXGPLTODNUVGFL-JZFBHDEDSA-N 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 230000004850 protein–protein interaction Effects 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 229960005206 pyrazinamide Drugs 0.000 description 1
- IPEHBUMCGVEMRF-UHFFFAOYSA-N pyrazinecarboxamide Chemical compound NC(=O)C1=CN=CC=N1 IPEHBUMCGVEMRF-UHFFFAOYSA-N 0.000 description 1
- VKNSAVOURPMBRN-UHFFFAOYSA-N pyridin-3-ylmethyl 2-[4-[(4-chlorophenyl)methyl]phenoxy]-2-methylbutanoate Chemical compound C=1C=CN=CC=1COC(=O)C(C)(CC)OC(C=C1)=CC=C1CC1=CC=C(Cl)C=C1 VKNSAVOURPMBRN-UHFFFAOYSA-N 0.000 description 1
- 229960003811 pyrithione disulfide Drugs 0.000 description 1
- 229960000948 quinine Drugs 0.000 description 1
- 229960005038 quinisocaine Drugs 0.000 description 1
- YREYEVIYCVEVJK-UHFFFAOYSA-N rabeprazole Chemical compound COCCCOC1=CC=NC(CS(=O)C=2NC3=CC=CC=C3N=2)=C1C YREYEVIYCVEVJK-UHFFFAOYSA-N 0.000 description 1
- YJQZYXCXBBCEAQ-UHFFFAOYSA-N ractopamine Chemical compound C=1C=C(O)C=CC=1C(O)CNC(C)CCC1=CC=C(O)C=C1 YJQZYXCXBBCEAQ-UHFFFAOYSA-N 0.000 description 1
- 239000012857 radioactive material Substances 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 229920013730 reactive polymer Polymers 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229940107685 reopro Drugs 0.000 description 1
- 125000006853 reporter group Chemical group 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002207 retinal effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- 108020004418 ribosomal RNA Proteins 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 229960003522 roquinimex Drugs 0.000 description 1
- AHTFMWCHTGEJHA-UHFFFAOYSA-N s-(2,5-dioxooxolan-3-yl) ethanethioate Chemical compound CC(=O)SC1CC(=O)OC1=O AHTFMWCHTGEJHA-UHFFFAOYSA-N 0.000 description 1
- QXRUCDDVUMKOPI-UHFFFAOYSA-N s-[2-(dimethylamino)ethyl] 9,9-dimethylacridine-10-carbothioate Chemical compound C1=CC=C2N(C(=O)SCCN(C)C)C3=CC=CC=C3C(C)(C)C2=C1 QXRUCDDVUMKOPI-UHFFFAOYSA-N 0.000 description 1
- WEXRVFDKZKSKQS-UHFFFAOYSA-N s-[2-(dipropylamino)ethyl] 2,2-diphenylethanethioate Chemical compound C=1C=CC=CC=1C(C(=O)SCCN(CCC)CCC)C1=CC=CC=C1 WEXRVFDKZKSKQS-UHFFFAOYSA-N 0.000 description 1
- DUTQZUMFFDWHBC-UHFFFAOYSA-N s-[2-[2-[(2-methoxyphenoxy)methyl]-1,3-thiazolidin-3-yl]-2-oxoethyl] ethanethioate Chemical compound COC1=CC=CC=C1OCC1N(C(=O)CSC(C)=O)CCS1 DUTQZUMFFDWHBC-UHFFFAOYSA-N 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 150000003902 salicylic acid esters Chemical class 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 108010073863 saruplase Proteins 0.000 description 1
- 210000004116 schwann cell Anatomy 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 229960003141 secobarbital sodium Drugs 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229940125723 sedative agent Drugs 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 150000004756 silanes Chemical class 0.000 description 1
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 239000003998 snake venom Substances 0.000 description 1
- 229940083542 sodium Drugs 0.000 description 1
- HELHAJAZNSDZJO-OLXYHTOASA-L sodium L-tartrate Chemical compound [Na+].[Na+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O HELHAJAZNSDZJO-OLXYHTOASA-L 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 239000001433 sodium tartrate Substances 0.000 description 1
- 229960002167 sodium tartrate Drugs 0.000 description 1
- 235000011004 sodium tartrates Nutrition 0.000 description 1
- PKWKMKHLVNZNDO-BLDYCALRSA-M sodium;(2r,3s,4r,5r)-2,3,4,5,6-pentahydroxy-1-[4-(hydroxymethylsulfamoyl)anilino]hexane-1-sulfonate Chemical compound [Na+].OCNS(=O)(=O)C1=CC=C(NC([C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO)S([O-])(=O)=O)C=C1 PKWKMKHLVNZNDO-BLDYCALRSA-M 0.000 description 1
- ULARYIUTHAWJMU-UHFFFAOYSA-M sodium;1-[4-(2,5-dioxopyrrol-1-yl)butanoyloxy]-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O ULARYIUTHAWJMU-UHFFFAOYSA-M 0.000 description 1
- VUFNRPJNRFOTGK-UHFFFAOYSA-M sodium;1-[4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexanecarbonyl]oxy-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)C1CCC(CN2C(C=CC2=O)=O)CC1 VUFNRPJNRFOTGK-UHFFFAOYSA-M 0.000 description 1
- JJGWLCLUQNFDIS-GTSONSFRSA-M sodium;1-[6-[5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]hexanoyloxy]-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)CCCCCNC(=O)CCCC[C@H]1[C@H]2NC(=O)N[C@H]2CS1 JJGWLCLUQNFDIS-GTSONSFRSA-M 0.000 description 1
- FCZYGJBVLGLYQU-UHFFFAOYSA-M sodium;2-[2-[2-[4-(2,4,4-trimethylpentan-2-yl)phenoxy]ethoxy]ethoxy]ethanesulfonate Chemical compound [Na+].CC(C)(C)CC(C)(C)C1=CC=C(OCCOCCOCCS([O-])(=O)=O)C=C1 FCZYGJBVLGLYQU-UHFFFAOYSA-M 0.000 description 1
- BNHGKKNINBGEQL-UHFFFAOYSA-M sodium;5-ethyl-5-(3-methylbutyl)pyrimidin-3-ide-2,4,6-trione Chemical compound [Na+].CC(C)CCC1(CC)C(=O)NC(=O)[N-]C1=O BNHGKKNINBGEQL-UHFFFAOYSA-M 0.000 description 1
- AXXJTNXVUHVOJW-UHFFFAOYSA-M sodium;5-pentan-2-yl-5-prop-2-enylpyrimidin-3-ide-2,4,6-trione Chemical compound [Na+].CCCC(C)C1(CC=C)C(=O)NC(=O)[N-]C1=O AXXJTNXVUHVOJW-UHFFFAOYSA-M 0.000 description 1
- CGTMEHPODKVIII-UHFFFAOYSA-M sodium;[(1,5-dimethyl-3-oxo-2-phenylpyrazol-4-yl)-(2-methylpropyl)amino]methanesulfonate Chemical compound [Na+].O=C1C(N(CS([O-])(=O)=O)CC(C)C)=C(C)N(C)N1C1=CC=CC=C1 CGTMEHPODKVIII-UHFFFAOYSA-M 0.000 description 1
- FNXKBSAUKFCXIK-UHFFFAOYSA-M sodium;hydrogen carbonate;8-hydroxy-7-iodoquinoline-5-sulfonic acid Chemical compound [Na+].OC([O-])=O.C1=CN=C2C(O)=C(I)C=C(S(O)(=O)=O)C2=C1 FNXKBSAUKFCXIK-UHFFFAOYSA-M 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 229950004330 spiroplatin Drugs 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 108010042747 stallimycin Proteins 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- WNIFXKPDILJURQ-UHFFFAOYSA-N stearyl glycyrrhizinate Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C)CCC(C(=O)OCCCCCCCCCCCCCCCCCC)(C)CC5C4=CC(=O)C3C21C WNIFXKPDILJURQ-UHFFFAOYSA-N 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 150000008143 steroidal glycosides Chemical class 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- QTENRWWVYAAPBI-YCRXJPFRSA-N streptomycin sulfate Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](N=C(N)N)[C@H](O)[C@@H](N=C(N)N)[C@H](O)[C@H]1O.CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](N=C(N)N)[C@H](O)[C@@H](N=C(N)N)[C@H](O)[C@H]1O QTENRWWVYAAPBI-YCRXJPFRSA-N 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 1
- 229940120904 succinylcholine chloride Drugs 0.000 description 1
- YOEWQQVKRJEPAE-UHFFFAOYSA-L succinylcholine chloride (anhydrous) Chemical compound [Cl-].[Cl-].C[N+](C)(C)CCOC(=O)CCC(=O)OCC[N+](C)(C)C YOEWQQVKRJEPAE-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000003774 sulfhydryl reagent Substances 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003457 sulfones Chemical group 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical class ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 229960004000 talbutal Drugs 0.000 description 1
- BJVVMKUXKQHWJK-UHFFFAOYSA-N talbutal Chemical compound CCC(C)C1(CC=C)C(=O)NC(=O)NC1=O BJVVMKUXKQHWJK-UHFFFAOYSA-N 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229960003188 temazepam Drugs 0.000 description 1
- QPWVHJDIDXILDG-SSUKDTCJSA-N tert-butyl 2-[2-[(2s,3s)-3-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-2-methyl-4-oxoazetidin-1-yl]oxyacetyl]oxyacetate Chemical compound C=1SC(N)=NC=1C(=N/OC)/C(=O)N[C@H]1[C@H](C)N(OCC(=O)OCC(=O)OC(C)(C)C)C1=O QPWVHJDIDXILDG-SSUKDTCJSA-N 0.000 description 1
- CNALVHVMBXLLIY-IUCAKERBSA-N tert-butyl n-[(3s,5s)-5-methylpiperidin-3-yl]carbamate Chemical compound C[C@@H]1CNC[C@@H](NC(=O)OC(C)(C)C)C1 CNALVHVMBXLLIY-IUCAKERBSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960002372 tetracaine Drugs 0.000 description 1
- GKCBAIGFKIBETG-UHFFFAOYSA-N tetracaine Chemical compound CCCCNC1=CC=C(C(=O)OCCN(C)C)C=C1 GKCBAIGFKIBETG-UHFFFAOYSA-N 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- WQVCHXLTSYVIPX-UHFFFAOYSA-J tetrasodium;2-[bis[hydroxy(oxido)phosphoryl]methyl]butanedioate Chemical compound [Na+].[Na+].[Na+].[Na+].OP(O)(=O)C(P([O-])([O-])=O)C(C([O-])=O)CC([O-])=O WQVCHXLTSYVIPX-UHFFFAOYSA-J 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- YXVCLPJQTZXJLH-UHFFFAOYSA-N thiamine(1+) diphosphate chloride Chemical compound [Cl-].CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N YXVCLPJQTZXJLH-UHFFFAOYSA-N 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- CWERGRDVMFNCDR-UHFFFAOYSA-M thioglycolate(1-) Chemical compound [O-]C(=O)CS CWERGRDVMFNCDR-UHFFFAOYSA-M 0.000 description 1
- ATGUDZODTABURZ-UHFFFAOYSA-N thiolan-2-ylideneazanium;chloride Chemical compound Cl.N=C1CCCS1 ATGUDZODTABURZ-UHFFFAOYSA-N 0.000 description 1
- 229960003279 thiopental Drugs 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 150000003585 thioureas Chemical class 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- RZWIIPASKMUIAC-VQTJNVASSA-N thromboxane Chemical compound CCCCCCCC[C@H]1OCCC[C@@H]1CCCCCCC RZWIIPASKMUIAC-VQTJNVASSA-N 0.000 description 1
- 229960000187 tissue plasminogen activator Drugs 0.000 description 1
- 229960001017 tolmetin Drugs 0.000 description 1
- UPSPUYADGBWSHF-UHFFFAOYSA-N tolmetin Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=C(CC(O)=O)N1C UPSPUYADGBWSHF-UHFFFAOYSA-N 0.000 description 1
- PHTUQLWOUWZIMZ-GZTJUZNOSA-N trans-dothiepin Chemical compound C1SC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 PHTUQLWOUWZIMZ-GZTJUZNOSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 102000003601 transglutaminase Human genes 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 230000007723 transport mechanism Effects 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 229940126307 triamcinolone acetate Drugs 0.000 description 1
- JOFWLTCLBGQGBO-UHFFFAOYSA-N triazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1Cl JOFWLTCLBGQGBO-UHFFFAOYSA-N 0.000 description 1
- 229960003386 triazolam Drugs 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 235000013337 tricalcium citrate Nutrition 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- HLXQFVXURMXRPU-UHFFFAOYSA-L trimethyl-[10-(trimethylazaniumyl)decyl]azanium;dibromide Chemical compound [Br-].[Br-].C[N+](C)(C)CCCCCCCCCC[N+](C)(C)C HLXQFVXURMXRPU-UHFFFAOYSA-L 0.000 description 1
- UZOHFHFMSPULPR-UHFFFAOYSA-L trimethyl-[2-(1,1,6-trimethylpiperidin-1-ium-2-carbonyl)oxyethyl]azanium;diiodide Chemical compound [I-].[I-].CC1CCCC(C(=O)OCC[N+](C)(C)C)[N+]1(C)C UZOHFHFMSPULPR-UHFFFAOYSA-L 0.000 description 1
- GQMVAUFIUVHMBB-UHFFFAOYSA-K trinaphthalen-2-yloxybismuthane Chemical compound C1=CC=CC2=CC(O[Bi](OC=3C=C4C=CC=CC4=CC=3)OC=3C=C4C=CC=CC4=CC=3)=CC=C21 GQMVAUFIUVHMBB-UHFFFAOYSA-K 0.000 description 1
- KZNBHWLDPGWJMM-UHFFFAOYSA-J trisodium;dioxido-oxo-sulfanylidene-$l^{6}-sulfane;gold(1+);dihydrate Chemical compound O.O.[Na+].[Na+].[Na+].[Au+].[O-]S([O-])(=O)=S.[O-]S([O-])(=O)=S KZNBHWLDPGWJMM-UHFFFAOYSA-J 0.000 description 1
- 229960004799 tryptophan Drugs 0.000 description 1
- 125000000430 tryptophan group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 229960002655 tubocurarine chloride Drugs 0.000 description 1
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- AQLJVWUFPCUVLO-UHFFFAOYSA-N urea hydrogen peroxide Chemical compound OO.NC(N)=O AQLJVWUFPCUVLO-UHFFFAOYSA-N 0.000 description 1
- VEPSYABRBFXYIB-PWXDFCLTSA-M vecuronium bromide Chemical compound [Br-].N1([C@@H]2[C@@H](OC(C)=O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)C)[N+]2(C)CCCCC2)CCCCC1 VEPSYABRBFXYIB-PWXDFCLTSA-M 0.000 description 1
- 229960004298 vecuronium bromide Drugs 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- NCYCYZXNIZJOKI-UHFFFAOYSA-N vitamin A aldehyde Natural products O=CC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-UHFFFAOYSA-N 0.000 description 1
- MECHNRXZTMCUDQ-RKHKHRCZSA-N vitamin D2 Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)/C=C/[C@H](C)C(C)C)=C\C=C1\C[C@@H](O)CCC1=C MECHNRXZTMCUDQ-RKHKHRCZSA-N 0.000 description 1
- 235000001892 vitamin D2 Nutrition 0.000 description 1
- 239000011653 vitamin D2 Substances 0.000 description 1
- 102100036537 von Willebrand factor Human genes 0.000 description 1
- 229960001134 von willebrand factor Drugs 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- HLDCSYXMVXILQC-UHFFFAOYSA-N xenysalate Chemical compound CCN(CC)CCOC(=O)C1=CC=CC(C=2C=CC=CC=2)=C1O HLDCSYXMVXILQC-UHFFFAOYSA-N 0.000 description 1
- 229960003434 xenysalate Drugs 0.000 description 1
- 229960000523 zalcitabine Drugs 0.000 description 1
- 229960002555 zidovudine Drugs 0.000 description 1
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 1
- WFPIAZLQTJBIFN-DVZOWYKESA-N zuclopenthixol Chemical compound C1CN(CCO)CCN1CC\C=C\1C2=CC(Cl)=CC=C2SC2=CC=CC=C2/1 WFPIAZLQTJBIFN-DVZOWYKESA-N 0.000 description 1
- WHNFPRLDDSXQCL-UAZQEYIDSA-N α-msh Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(N)=O)NC(=O)[C@H](CO)NC(C)=O)C1=CC=C(O)C=C1 WHNFPRLDDSXQCL-UAZQEYIDSA-N 0.000 description 1
- OENHQHLEOONYIE-JLTXGRSLSA-N β-Carotene Chemical compound CC=1CCCC(C)(C)C=1\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C OENHQHLEOONYIE-JLTXGRSLSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/12—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by a special physical form, e.g. emulsion, microcapsules, liposomes, characterized by a special physical form, e.g. emulsions, dispersions, microcapsules
- A61K51/1241—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by a special physical form, e.g. emulsion, microcapsules, liposomes, characterized by a special physical form, e.g. emulsions, dispersions, microcapsules particles, powders, lyophilizates, adsorbates, e.g. polymers or resins for adsorption or ion-exchange resins
- A61K51/1255—Granulates, agglomerates, microspheres
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/542—Carboxylic acids, e.g. a fatty acid or an amino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6921—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere
- A61K47/6925—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a microcapsule, nanocapsule, microbubble or nanobubble
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/0002—General or multifunctional contrast agents, e.g. chelated agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/0019—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules
- A61K49/0021—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules the fluorescent group being a small organic molecule
- A61K49/0041—Xanthene dyes, used in vivo, e.g. administered to a mice, e.g. rhodamines, rose Bengal
- A61K49/0043—Fluorescein, used in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/0019—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules
- A61K49/0045—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules the fluorescent agent being a peptide or protein used for imaging or diagnosis in vivo
- A61K49/0047—Green fluorescent protein [GFP]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/005—Fluorescence in vivo characterised by the carrier molecule carrying the fluorescent agent
- A61K49/0054—Macromolecular compounds, i.e. oligomers, polymers, dendrimers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/005—Fluorescence in vivo characterised by the carrier molecule carrying the fluorescent agent
- A61K49/0056—Peptides, proteins, polyamino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/005—Fluorescence in vivo characterised by the carrier molecule carrying the fluorescent agent
- A61K49/0058—Antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0063—Preparation for luminescence or biological staining characterised by a special physical or galenical form, e.g. emulsions, microspheres
- A61K49/0069—Preparation for luminescence or biological staining characterised by a special physical or galenical form, e.g. emulsions, microspheres the agent being in a particular physical galenical form
- A61K49/0089—Particulate, powder, adsorbate, bead, sphere
- A61K49/0091—Microparticle, microcapsule, microbubble, microsphere, microbead, i.e. having a size or diameter higher or equal to 1 micrometer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/22—Echographic preparations; Ultrasound imaging preparations ; Optoacoustic imaging preparations
- A61K49/222—Echographic preparations; Ultrasound imaging preparations ; Optoacoustic imaging preparations characterised by a special physical form, e.g. emulsions, liposomes
- A61K49/223—Microbubbles, hollow microspheres, free gas bubbles, gas microspheres
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/06—Macromolecular compounds, carriers being organic macromolecular compounds, i.e. organic oligomeric, polymeric, dendrimeric molecules
- A61K51/065—Macromolecular compounds, carriers being organic macromolecular compounds, i.e. organic oligomeric, polymeric, dendrimeric molecules conjugates with carriers being macromolecules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/12—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by a special physical form, e.g. emulsion, microcapsules, liposomes, characterized by a special physical form, e.g. emulsions, dispersions, microcapsules
- A61K51/1217—Dispersions, suspensions, colloids, emulsions, e.g. perfluorinated emulsion, sols
- A61K51/1227—Micelles, e.g. phospholipidic or polymeric micelles
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
- Y10S977/927—Diagnostic contrast agent
- Y10S977/928—X-ray agent
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
- Y10S977/927—Diagnostic contrast agent
- Y10S977/929—Ultrasound contrast agent
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
- Y10S977/927—Diagnostic contrast agent
- Y10S977/93—MRI contrast agent
Definitions
- This invention relates to diagnostic and/or therapeutically active agents, more particularly to diagnostic and/or therapeutically active agents incorporating moieties having affinity for sites and/or structures within the body so that diagnostic imaging and/or therapy of particular locations within the body may be enhanced.
- diagnostic agents for use in ultrasound imaging which are hereinafter referred to as targeted ultrasound contrast agents.
- ultrasonic imaging comprises a potentially valuable diagnostic tool, for example in studies of the vascular system, particularly in cardiography, and of tissue microvasculature.
- contrast agents has been proposed to enhance the acoustic images so obtained, including suspensions of solid particles, emulsified liquid droplets, gas bubbles and encapsulated gases or liquids. It is generally accepted that low density contrast agents which are easily compressible are particularly efficient in terms of the acoustic backscatter they generate, and considerable interest has therefore been shown in the preparation of gas-containing and gas-generating systems.
- Gas-containing contrast media are also known to be effective in magnetic resonance (MR) imaging, e.g. as susceptibility contrast agents which will act to reduce MR signal intensity.
- Oxygen-containing contrast media also represent potentially useful paramagnetic MR contrast agents.
- gases such as carbon dioxide may be used as negative oral contrast agents or intravascular contrast agents.
- radioactive gases e.g. radioactive isotopes of inert gases such as xenon
- scintigraphy for example for blood pool imaging.
- Targeted ultrasound contrast agents may be regarded as comprising (i) a reporter moiety capable of interacting with ultrasound irradiation to generate a detectable signal; (ii) one or more vectors having affinity for particular target sites and/or structures within the body, e.g. for specific cells or areas of pathology; and (iii) one or more linkers connecting said reporter and vector(s), in the event that these are not directly joined.
- the molecules and/or structure to which the contrast agent is intended to bind will hereinafter be referred to as the target.
- the target In order to obtain specific imaging of a selected region/structure in the body the target must be present and available in this region/structure. Ideally it will be expressed only in the region of interest, but usually will also be present at other locations in the body, creating possible background problems.
- the target may either be a defined molecular species (i.e. a target molecule) or an unknown molecule or more complex structure (i.e. a target structure) which is present in the area to be imaged, and is able to bind specifically or selectively to a given vector molecule.
- the vector is attached to the reporter moiety in order to bind these moieties to the region/structure to be imaged.
- the vector may bind specifically to a chosen target, or it may bind only selectively, having affinty also for a limited number of other molecules/structures, again creating possible background problems.
- U.S. Pat No. 5,531,980 is directed to systems in which the reporter comprises an aqueous suspension of air or gas microbubbles stabilised by one or more film-forming surfactants present at least partially in lamellar or laminar form, said surfactant(s) being bound to one or more vectors comprising “bioactive species designed for specific targeting purposes”. It is stated that the microbubbles are not directly encapsulated by surfactant material but rather that this is incorporated in liquid-filled liposomes which stabilise the microbubbles.
- lamellar or laminar surfactant material such as phospholipids present in such liposomes will inevitably be present in the form of one or more lipid bilayers with the lipophilic tails “back-to-back” and the hydrophilic heads both inside and outside (see e.g. Schneider, M. on “Liposomes as drug carriers: 10 years of research” in Drug targeting, Nyon, Switzerland , 3-5 Oct . 1984, Buri, P. and Gumma, A. (Ed), Elsevier, Amsterdam 1984).
- EP-A-0727225 describes targeted ultrasound contrast agents in which the reporter comprises a chemical having a sufficient vapour pressure such that a proportion of it is a gas at the body temperature of the subject.
- This chemical is associated with a surfactant or albumin carrier which includes a protein-, peptide- or carbohydrate-based cell adhesion molecule ligand as vector.
- the receptor moieties in such contrast agents correspond to the phase shift colloid systems described in WO-A-9416739; it is now recognised that administration of such phase shift colloids may lead to generation of microbubbles which grow uncontrollably, possibly to the extent where they cause potentially dangerous embolisation of, for example, the myocardial vasculature and brain (see e.g. Schwarz, Advances in Echo - Contrast [ 1994(3)], pp 48-49).
- WO-A-9320802 proposes that tissue-specific ultrasonic image enhancement may be achieved using acoustically reflective oligolamellar liposomes conjugated to tissue-specific ligands such as antibodies, peptides, lectins etc.
- tissue-specific ligands such as antibodies, peptides, lectins etc.
- the liposomes are deliberately chosen to be devoid of gas and so will not have the advantageous echogenic properties of gas-based ultrasound contrast agents.
- Further references to this technology e.g. in targeting to fibrin, thrombi and atherosclerotic areas are found in publications by Alkanonyuksel, H. et al. in J. Pharm. Sci . (1996) 85(5), 486-490 ; J. Am. Coll. Cardiol . (1996) 27(2) Suppl A, 298A; and Circulation , 68 Sci. Sessions , Anaheim 13-16 Nov. 1995.
- these prior art targeted contrast agents are intended to enhance contrast at specific sites in the body, for example tumour cells, by using one vector to bind strongly to one target, in order to achieve concentration at the target cells.
- the present invention is based in part on the finding that diagnostic and/or therapeutically active agents with more favourable properties may be obtained by use of multiple kinds of vector-target interactions (e.g. involving agents associated with a plurality of different vectors and/or with one or more vectors having affinity for different targets on the same or different cell types).
- binding of gas-containing and gas-generating diagnostic and/or therapeutic agents may, for example, be obtained by forming multiple binding pairs between one vector with specificity for more than one receptor or between more than one vector with affinity for one or more types of target, with either low or high affinities.
- Such multiple binding of the vector-conjugated agent to one or more target molecules/structures may result in advantageous targeting properties, for example by enhancing target specificity and/or by distinguishing interactions at a desired target area from background interactions with lower levels of molecules/structures similar to target expressed elsewhere in the body.
- the present invention is based on the finding that the desired binding of gas-containing and gas-generating diagnostic and/or therapeutic agents may be obtained by forming multiple binding pairs with low affinity between one type of vector and one type of target, or by forming multiple binding pairs between one or more types of vectors and one or more types of target, with either low or high affinities.
- multiple binding of the vector conjugated agent to one or more target molecules/structures may have advantageous targeting properties, for example in enhancing target specificity and/or in distinguishing interactions at a desired target area from background interactions with lower levels of molecules/structures similar to target expressed elsewhere in the body.
- a targetable diagnostic and/or therapeutically active agent e.g. an ultrasound contrast agent
- a suspension in an aqueous carrier liquid e.g. an injectable carrier liquid
- a reporter comprising gas-containing or gas-generating material characterised in that said agent is capable of forming at least two types of binding pairs, e.g. being conjugated to at least two vectors or to one vector capable of binding to at least two binding sites.
- One advantageous embodiment of the invention is based on the additional finding that limited adhesion to targets is a highly useful property of diagnostic and/or therapeutically active agents, which property may be achieved using vectors giving temporary retention rather than fixed adhesion to a target.
- agents rather than being fixedly retained at specific sites, may for example effectively exhibit a form of retarded flow along the vascular endothelium by virtue of their transient interactions with endothelial cells.
- Such agents may thus become concentrated on the walls of blood vessels, in the case of ultrasound contrast agents providing enhanced echogenicity thereof relative to the bulk of the bloodstream, which is devoid of anatomical features.
- the present invention is well suited to imaging changes occurring in normal blood vessels which are situated in areas of tissue necrosis.
- binding affinities are dependent on numbers of interactions as well as their strength.
- the density of vector molecules at the surface of the reporter units may therefore be selected so as appropriately to adjust the degree of interactions between particular agents and targets.
- multiple-specificity is also used to describe an injectable carrier liquid, of gas-containing or gas-generating material composed of one or more vectors with a specificity for one or more cellular surface receptors while at the same time comprising a second element with specificity for a substrate or receptor system binding to which induces a therapeutic response.
- multiple-specific imaging agents comprising a targeting vector, such as the anti-fibrin antibody described by Lanza et al.
- annexin V atherosclerotic plaque binding peptides such as YRALVDTLK, or any other vector known to associate with fibrin clots, in combination with a drug or enzyme with fibrinolytic activity such as streptokinase, plasminogen activator (tPA), urokinase (uPA) or prourokinase (scuPA) resulting in a localised therapeutic antithrombotic effect.
- tPA plasminogen activator
- uPA urokinase
- scuPA prourokinase
- reporter carrying a multiplicity of vectors with relatively low affinity for the target may be advantageous in this situation, since the reporter will then tend to concentrate in regions of high target density which permit multiple (and therefore strong) binding to the reporter (e.g. a gas-containing agent incorporating the vectors folic acid and glutathione for multiple-specific binding to folic acid receptors and glutathione-S-trasferase receptors respectively which are over-expressed as tumour cells).
- low affinity vectors may be regarded as having an association constant Kafor interaction with a target molecule or structure of less than 10 8 M ⁇ 1 , e.g. less than 10 7 M ⁇ 1 , preferably less than 10 6 M ⁇ 1 .
- Kafor interaction with a target molecule or structure of less than 10 8 M ⁇ 1 , e.g. less than 10 7 M ⁇ 1 , preferably less than 10 6 M ⁇ 1 .
- a further embodiment of this invention is thus based on the finding that the desired binding of gas-containing and gas-generating diagnostic and/or therapeutic agents may be obtained by forming binding pairs with low affinity between more than one type of vector and one or more type of target. Multiple vectors may therfore be used to increase specificity, so that the reporter will bind only to target cells or structures expressing a particular combination of target molecules.
- Products comprising two or more vectors with different specificities, i.e. which bind to different target molecules on different cells, may advantageously be used as “general purpose” agents for detection of a range of diseases, e.g. different forms of cancer.
- the use of such agents may enable detection of metastases, which are often heterogeneous with respect to expression of target molecules (i.e. antigens).
- the reporter unit will usually remain attached to the vectors.
- the vector (often, a monoclonal antibody) is administered alone; subsequently, the reporter is administered, coupled to a moiety which is capable of specifically binding the vector molecule (when the vector is an antibody, the reporter may be coupled to an immunoglobulin-binding molecule, such as protein A or an anti-immunoglobulin antibody).
- an advantage of this protocol is that time may be allowed for elimination of the vector molecules that do not bind their targets, substantially reducing the background problems that are connected with the presence of an excess of reporter-vector conjugate.
- pre-targeting with one specific vector might be envisaged, followed by reporter units that are coupled to another vector and a moiety which binds the first vector.
- Vectors useful in accordance with the invention include ligands for cell adhesion proteins, as well as cell adhesion proteins themselves where these have corresponding ligands on endothelial cell surfaces.
- cell adhesion proteins include integrins, most of which bind the Arg-Gly-Asp (RGD) amino acid sequence.
- the vector may be targeted to specific cell adhesion proteins expressed mainly on activated endothelial cells such as are found at or close to sites of inflammation or other pathological responses.
- Other vectors which may be used include proteins and peptides that bind to cell-surface proteoglycans, which are complexes of proteins and sulphated polysaccarides found on most cells, including endothelial cells.
- proteoglycans contribute to the negative surface charge of all nucleated cells from vertebrate animals; this charge may also be exploited in accordance with the invention by using positively charged vectors, e.g. comprising cationic lipids, which will interact electrostatically with the endothelial surface.
- a further aspect of the present invention is for example where a vector or vectors is attached to the reporter or included non-covalently into the reporter in a manner where the said vector or vectors is not readily exposed to the targets or receptors.
- Increased tissue specificity may therefore be achieved by applying an additional process to expose the vectors, e.g. the agent is exposed after administration to external ultrasound to change the diffusibility of the moieties containing the vectors.
- the reporter may be in any convenient form, for example being any appropriate gas-containing or gas-generating ultrasound contrast agent formulation.
- Representative examples of such formulations include microbubbles of gas stabilised (e.g. at least partially encapsulated) by a coalescence-resistant surface membrane (for example gelatin, e.g. as described in WO-A-8002365), a filmogenic protein (for example an albumin such as human serum albumin, e.g. as described in U.S. Pat No. 4,718,433, U.S. Pat No. 4,774,958, U.S. Pat No.
- gas-containing contrast agent formulations include gas-containing solid systems, for example microparticles (especially aggregates of microparticles) having gas contained therewithin or otherwise associated therewith (for example being adsorbed on the surface thereof and/or contained within voids, cavities or pores therein, e.g. as described in EP-A-0122624, EP-A-0123235, EP-A-0365467, WO-A-9221382, WO-A-9300930, WO-A-9313802, WO-A-9313808 or WO-A-9313809).
- the echogenicity of such microparticulate contrast agents may derive directly from the contained/associated gas and/or from gas (e.g. microbubbles) liberated from the solid material (e.g. upon dissolution of the microparticulate structure).
- Gas microbubbles and other gas-containing materials such as microparticles preferably have an initial average size not exceeding 10 ⁇ m (e.g. of 7 ⁇ m or less) in order to permit their free passage through the pulmonary system following administration, e.g. by intravenous injection.
- phospholipid-containing compositions are employed in accordance with the invention, e.g. in the form of phospholipid-stabilised gas microbubbles
- useful phospholipids include lecithins (i.e. phosphatidylcholines), for example natural lecithins such as egg yolk lecithin or soya bean lecithin and synthetic or semisynthetic lecithins such as dimyristoylphosphatidylcholine, dipalmitoylphosphatidylcholine or distearoylphosphatidylcholine; phosphatidic acids; phosphatidylethanolamines; phosphatidylserines; phosphatidylglycerols; phosphatidylinositols; cardiolipins; sphingomyelins; fluorinated analogues of any of the foregoing; mixtures of any of the foregoing and mixtures with other lipids such as cholesterol.
- lecithins
- phospholipids predominantly (e.g. at least 75%) comprising molecules individually bearing net overall charge, e.g. negative charge, for example as in naturally occurring (e.g. soya bean or egg yolk derived), semisynthetic (e.g. partially or fully hydrogenated) and synthetic phosphatidylserines, phosphatidylglycerols, phosphatidylinositols, phosphatidic acids and/or cardiolipins, may be particularly advantageous.
- naturally occurring e.g. soya bean or egg yolk derived
- semisynthetic e.g. partially or fully hydrogenated
- synthetic phosphatidylserines e.g. partially or fully hydrogenated
- phosphatidylglycerols phosphatidylglycerols
- phosphatidylinositols phosphatidic acids and/or cardiolipins
- exemplary lipids which may be used to prepare gas-containing contrast agents include fatty acids, stearic acid, palmitic acid, 2-n-hexadecylstearic acid, oleic acid and other acid containing lipid structures. These lipid structures are considered particularly interesting when coupled by amide bond formation to amino acids containing one or more amino groups.
- the resulting lipid modified amino acids e.g. dipalmitoyllysine, distearoyl-2,3-diaminopropionic acid
- a further extension of this invention relates to the synthesis of lipopeptide structures comprising a lipid reporter attached to a linker portion (e.g. PEG, polyamino acid, alkylhalide etc) the said linker being suitably functionalised for coupling to one or more vector molecules.
- a linker portion e.g. PEG, polyamino acid, alkylhalide etc
- the said linker being suitably functionalised for coupling to one or more vector molecules.
- a positively charged linker element e.g. two or more lysine residues
- Multiple-specific targeting is achievable by mixing and ‘doping’ of phospholipid gas-containing structures with one or more targeted lipopeptide sequences.
- Multiple-specificity can also be achieved by assembling more than one vector on a branched lysine core structure such as those described by Tam et. al. (Proc. Natl. Acad. Sci. USA, 1989, 86, 9084) or by incorporating multiple vectors in a linear sequence. Multiple-specificity can also be achieved using lipopeptides or phospholipids comprising combinatorial libraries synthesised by chemical synthesis as described by Lowe (Combinatorial Chemistry, Chemical Society Reviews, 1995, 309-317).
- microbubbles carrying one or more reactive groups for non-specific reaction with receptor molecules located on cell surfaces.
- Microbubbles comprising a thiol moiety,for example, can bind to cell surface receptors via disulphide exchange reactions. The reversible nature of this covalent bond means that bubble flow can be controlled by altering the redox environment.
- activated microbubbles of membranes comprising active esters such as N-hydroxysuccinimide esters can be used to modify amino groups found on a multiplicity of cell surface molecules.
- gas-containing microparticulate materials which may be useful in accordance with the invention include carbohydrates (for example hexoses such as glucose, fructose or galactose; disaccharides such as sucrose, lactose or maltose; pentoses such as arabinose, xylose or ribose; ⁇ -, ⁇ - and ⁇ -cyclodextrins; polysaccharides such as starch, hydroxyethyl starch, amylose, amylopectin, glycogen, inulin, pulullan, dextran, carboxymethyl dextran, deXtran phosphate, ketodextran, aminoethyldextran, alginates, chitin, chitosan, hyaluronic acid or heparin; and sugar alcohols, including alditols such as mannitol or sorbitol), inorganic salts (e.g.
- X-ray contrast agents e.g. any of the commercially available carboxylic acid and non-ionic amide contrast agents typically containing at least one 2,4,6-triiodophenyl group having substituents such as carboxyl, carbamoyl, N-alkylcarbamoyl, N-hydroxyalkylcarbamoyl, acylamino, N-alkylacylamino or acylaminomethyl at the 3- and/or 5-positions, as in metrizoic acid, diatrizoic acid, iothalamic acid, ioxaglic acid, iohexol, iopentol, iopamidol, iodixanol, iopromide, metrizamide, iodipamide, meglumine iodipamide, meglumine acetrizoate and meglumine diatriz
- gas any biocompatible gas may be present in the reporter of contrast agents according to the invention, the term “gas” as used herein including any substances (including mixtures) substantially or completely in gaseous (including vapour) form at the normal human body temperature of 37° C.
- the gas may thus, for example, comprise air; nitrogen; oxygen; carbon dioxide; hydrogen; an inert gas such as helium, argon, xenon or krypton; a sulphur fluoride such as sulphur hexafluoride, disulphur decafluoride or trifluoromethylsulphur pentafluoride; selenium hexafluoride; an optionally halogenated silane such as methylsilane or dimethylsilane; a low molecular weight hydrocarbon (e.g.
- an alkane such as methane, ethane, a propane, a butane or a pentane, a cycloalkane such as cyclopropane, cyclobutane or cyclopentane, an alkene such as ethylene, propene, propadiene or a butene, or an alkyne such as acetylene or propyne; an ether such as dimethyl ether; a ketone; an ester; a halogenated low molecular weight hydrocarbon (e.g. containing up to 7 carbon atoms); or a mixture of any of the foregoing.
- an alkane such as methane, ethane, a propane, a butane or a pentane
- a cycloalkane such as cyclopropane, cyclobutane or cyclopentane
- an alkene such as ethylene, propene, propadiene or a butene
- biocompatible halogenated hydrocarbon gases may, for example, be selected from bromochlorodifluoromethane, chlorodifluoromethane, dichlorodifluoromethane, bromotrifluoromethane, chlorotrifluoromethane, chloropentafluoroethane, dichlorotetrafluoroethane, chlorotrifluoroethylene, fluoroethylene, ethylfluoride, 1,1-difluoroethane and perfluorocarbons, e.g.
- perfluoroalkanes such as perfluoromethane, perfluoroethane, perfluoropropanes, perfluorobutanes (e.g. perfluoro-n-butane, optionally in admixture with other isomers such as perfluoro-iso-butane), perfluoropentanes, perfluorohexanes and perfluoroheptanes; perfluoroalkenes such as perfluoropropene, perfluorobutenes-(e.g.
- halogenated gases include methyl chloride, fluorinated (e.g.
- perfluorinated ketones such as perfluoroacetone and fluorinated (e.g. perfluorinated) ethers such as perfluorodiethyl ether.
- perfluorinated gases for example sulphur hexafluoride and perfluorocarbons such as perfluoropropane, perfluorobutanes and perfluoropentanes, may be particularly advantageous in view of the recognised high stability in the bloodstream of microbubbles containing such gases.
- the reporter may be made by any convenient process, for example by making gas-containing or gas-generating formulations.
- Representative examples include the preparation of a suspension of gas microbubbles by contacting a surfactant with gas and mixing them in the presence of an aqueous carrier, as described in WO 9115244; or by atomising a solution or dispersion of a wall-forming material in the presence of a gas in order to obtain hollow microcapsules, as described in EP 512693A1; preparation of solid microspheres by a double emulsion process, as described in U.S. Pat. No.
- a suitable process for attachment of the desired vector to the reporter comprises a surface modification of the preformed reporter with a suitable linker employing reactive groups on the surface of both the reporter and vector. It may be particularly advantageous physically to mix the reporter material with the vector-containing substance at any step of the process. Such a process will result in incorporation or an attachment of the vector to the reporter. An optional process step may remove the excess of vector not bound to the reporter by washing the gas-containing particles following separation, by for example, floatation.
- a preferred aspect is the use of lipopeptide structures incorporating functional groups such as thiol, maleimide biotin etc. which can be premixed if desired with other reporter molecules before formation of gas-containing agents.
- the attachment of vector molecules may be carried out using the linker reagents listed below.
- Linking of a reporter unit to the desired vectors may be achieved by covalent or non-covalent means, usually involving interaction with one or more functional groups located on the reporter and/or vectors.
- functional groups located on the reporter and/or vectors.
- chemically reactive functional groups include amino, hydroxyl, sulfhydryl, carboxyl, and carbonyl groups, as well as carbohydrate groups, vicinal diols, thioethers, 2-aminoalcohols, 2-aminothiols, guanidinyl, imidazolyl and phenolic groups.
- Covalent coupling of reporter and vectors may therefore be effected using linking agents containing reactive moieties capable of reaction with such functional groups.
- N-Maleimide derivatives are also considered selective towards sulfhydryl groups, but may additionaly be useful in coupling to amino groups under certain conditions.
- N-maleimides may be incorporated into linking systems for reporter-vector conjugation as described by Kitagawa, T. et al. in Chem. Pharm. Bull . (1981) 29, 1130 or used as polymer crosslinkers for bubble stabilisation as described by Kovacic, P. et al. in J. Am. Chem. Soc . (1959) 81, 1887.
- Reagents such as 2-iminothiolane, e.g. as described by Traut, R. et al. in Biochemistry (1973) 12, 3266, which introduce a thiol group through conversion of an amino group, may be considered as sulfhydryl reagents if linking occurs through the formation of disulphide bridges.
- reagents which introduce reactive disulphide bonds into either the reporter or the vectors may be useful, since linking may be brought about by disulphide exchange between the vector and reporter;
- examples of such reagents include Ellman's reagent (DTNB), 4,4′-dithiodipyridine, methyl-3-nitro-2-pyridyl disulphide and methyl-2-pyridyl disulphide (described by Kimura, T. et al. in Analyt. Biochem . (1982) 122, 271).
- Examples of reactive moieties capable of reaction with amino groups include alkylating and acylating agents.
- Representative alkylating agents include:
- Representative amino-reactive acylating agents include:
- Carbonyl groups such as aldehyde functions may be reacted with weak protein bases at a pH such that nucleophilic protein side-chain functions are protonated.
- Weak bases include 1,2-aminothiols such as those found in N-terminal cysteine residues, which selectively form stable 5-membered thiazolidine rings with aldehyde groups, e.g. as described by Ratner, S. et al. in J. Am. Chem. Soc . (1937) 59, 200.
- Other weak bases such as phenyl hydrazones may be used, e.g. as described by Heitzman, H. et al. in Proc. Natl. Acad. Sci. USA (1974) 71, 3537.
- Aldehydes and ketones may also be reacted with amines to form Schiff's bases, which may advantageously be stabilised through reductive amination.
- Alkoxylamino moieties readily react with ketones and aldehydes to produce stable alkoxamines, e.g. as described by Webb, R. et al. in Bioconjugate Chem . (1990) 1, 96.
- reactive moieties capable of reaction with carboxyl groups include diazo compounds such as diazoacetate esters and diazoacetamides, which react with high specificity to generate ester groups, e.g. as described by Herriot R. M. in Adv. Protein Chem . (1947) 3, 169.
- Carboxylic acid modifying reagents such as carbodiimides, which react through O-acylurea formation followed by amide bond formation, may also usefully be employed; linking may be facilitated through addition of an amine or may result in direct vector-receptor coupling.
- Useful water soluble carbodiimides include 1-cyclohexyl-3-(2-morpholinyl-4-ethyl)carbodiimide (CMC) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), e.g. as described by Zot, H. G. and Puett, D. in J. Biol. Chem . (1989) 264, 15552.
- carboxylic acid modifying reagents include isoxazolium derivatives such as Wo6dwards reagent K; chloroformates such as p-nitrophenylchloroformate; carbonyldiimidazoles such as 1,1′-carbonyldiimidazole; and N-carbalkoxydihydroquinolines such as N-(ethoxycarbonyl)-2-ethoxy-1,2-dihydroquinoline.
- vicinal diones such as p-phenylenediglyoxal, which may be used to react with guanidinyl groups, e.g. as described by Wagner et al. in Nucleic acid Res . (1978) 5, 4065; and diazonium salts, which may undergo electrophilic substitution reactions, e.g. as described by Ishizaka, K. and Ishizaka T. in J. Immunol . (1960) 85, 163.
- Bis-diazonium compounds are readily prepared by treatment of aryl diamines with sodium nitrite in acidic solutions. It will be appreciated that functional groups in the reporter and/or vector may if desired be converted to other functional groups prior to reaction, e.g.
- Examples of methods useful for this purpose include conversion of amines to carboxylic acids using reagents such as dicarboxylic anhydrides; conversion of amines to thiols using reagents such as N-acetylhomocysteine thiolactone, S-acetylmercaptosuccinic anhydride, 2-iminothiolane or thiol-containing succinimidyl derivatives; conversion of thiols to carboxylic acids using reagents such as a-haloacetates; conversion of thiols to amines using reagents such as ethylenimine or 2-bromoethylamine; conversion of carboxylic acids to amines using reagents such as carbodiimides followed by diamines; and conversion of alcohols to thiols using reagents such as tosyl chloride followed by transesterification with thioacetate and hydrolysis to the thiol
- Vector-receptor coupling may also be effected using enzymes as zero-length crosslinking agents; thus, for example, transglutaminase, peroxidase and xanthine oxidase have been used to produce crosslinked products.
- Reverse proteolysis may also be used for crosslinking through amide bond formation.
- Non-covalent vector-receptor coupling may, for example, be effected by electrostatic charge interactions e.g. between a polylysinyl-functionalised reporter and a polyglutamyl-functionalised vector, through chelation in the form of stable metal complexes or through high affinity binding interaction such as avidin/biotin binding.
- Polylysine, coated non-covalently to the negatively charged membrane surface can also increase non-specifically the affinity of a microbubble for a cell through charge interactions.
- vectors may be coupled to a protein or peptide sequence known to bind phospholipids.
- a single molecule of phospholipid may attach to a protein such as a translocase, while other proteins may attach to surfaces consisting mainly of phospholipid head groups and so may be used to attach vectors to phospholipid microspheres;
- a protein such as a translocase
- other proteins may attach to surfaces consisting mainly of phospholipid head groups and so may be used to attach vectors to phospholipid microspheres;
- ⁇ 2-glycoprotein I Chon, A., Semple, S. C. and Cullis, P. R., Journal of Biological Chemistry (1995) 270, 25845-25849.
- Phosphatidylserine-binding proteins have been described, e.g. by Igarashi, K. et al.
- Annexins are a class of phospholipid-binding proteins, many of which bind particularly avidly to phosphatidyl-serine (reviewed in Raynal, P. and H. B. Pollard. Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins”. Biochim. Biophys. Acta 1197: 63-93).
- a conjugate of a vector with such a phosphatidylserine-binding protein may therefore be used to attach the vector to phosphatidylserine-encapsulated microbubbles.
- the amino acid sequence of a binding protein is known, the phospholipid-binding portion may be synthesised or isolated and used for conjugation with a vector, thus avoiding the biological activity which may be located elsewhere in the molecule.
- phage libraries displaying small peptides could be used for such selection.
- the selection may be made by simply mixing the microspheres and the phage display library and eluting the phages binding to the floating microspheres. If desired, the selection can be done under “physiological conditions” (e.g. in blood) to eliminate peptides which cross-react with blood components.
- physiological conditions e.g. in blood
- binding moieties identified in this way may be coupled (chemically via peptide synthesis, or at the DNA-level using recombinant vectors) to a vector molecule, constituting a general tool for attaching any vector molecule to the microspheres.
- a vector which comprises or is coupled to a peptide or lipopeptide linker which contains a element capable of mediating membrane insertion may also be useful.
- a peptide or lipopeptide linker which contains a element capable of mediating membrane insertion
- Non-bioactive molecules consisting of known membrane insertion anchor/signal groups may also be used as vectors for certain applications, an example being the H1 hydrophobic segment from the Na, K-ATPase ⁇ -subunit described by Xie, Y. and Morimoto, T. in J. Biol. Chem . (1995) 270(20), 11985-11991.
- the anchor group may also be fatty acid(s) or cholesterol.
- Coupling may also be effected using avidin or streptavidin, which have four high affinity binding sites for biotin. Avidin may therefore be used to conjugate vector to reporter if both vector and reporter are biotinylated. Examples are described by Bayer, E. A. and Wilchek, M. in Methods Biochem. Anal . (1980) 26, 1. This method may also be extended to include linking of reporter to reporter, a process which may encourage bubble association and consequent potentially increased echogenicity.
- Non-covalent coupling may also utilise the bifunctional nature of bispecific immunoglobulins. These molecules can specifically bind two antigens, thus linking them. For example, either bispecific IgG or chemically engineered bispecific F(ab)′2 fragments may be used as linking agents. Heterobifunctional bispecific antibodies have also been reported for linking two different antigens, e.g. as described by Bode, C. et al. in J. Biol. Chem . (1989) 264, 944 and by Staerz, U. D. et al. in Proc. Natl. Acad. Sci. USA (1986) 83, 1453. Similarly, any reporter and/or vector containing two or more antigenic determinants (e.g. as described by Chen, Aa et al. in Am. J. Pathol . (1988) 130, 216) may crosslink antibody molecules and lead to formation of multi-bubble cross-linked assemblies of potentially increased echogenicity.
- So-called zero-length linking agents which induce direct covalent joining of two reactive chemical groups without introducing additional linking material (e.g. as in amide bond formation induced using carbodiimides or enzymatically) may, if desired, be used, as may agents such as biotin/avidin systems which induce non-covalent reporter-vector linking and agents which induce hydrophobic or electrostatic interactions.
- the linking agent will comprise two or more reactive moieties, e.g. as described above, connected by a spacer element.
- a spacer element permits bifunctional linkers to react with specific functional groups within a molecule or between two different molecules, resulting in a bond between these two components and introducing extrinsic linker-derived material into the reporter-vector conjugate.
- the reactive moieties in a linking agent may be the same (homobifunctional agents) or different (heterobifunctional agents or, where several dissimilar reactive moieties are present, heteromultifunctional agents), providing a diversity of potential reagents that may bring about covalent bonding between any chemical species, either intramolecularly or intermolecularly.
- extrinsic material introduced by the linking agent may have a critical bearing on the targeting ability and general stability of the ultimate product.
- labile linkages e.g. containing spacer arms which are biodegradable or chemically sensitive or which incorporate enzymatic cleavage sites.
- the spacer may include polymeric components, e.g. to act as surfactants and enhance bubble stability.
- the spacer may also contain reactive moieties, e.g. as described above to enhance surface crosslinking, or it may contain a tracer element such as a fluorescent probe, spin label or radioactive material.
- Spacer elements may typically consist of aliphatic chains which effectively separate the reactive moieties of the linker by distances of between 5 and 30 ⁇ . They may also comprise macromolecular structures such as poly(ethylene glycols). Such polymeric structures, hereinafter referred to as PEGS, are simple, neutral polyethers which have been given much attention in biotechnical and biomedical applications (see e.g. Milton Harris, J. (ed) “ Poly ( ethylene glycol ) chemistry, biotechnical and biomedical applications ” Plenum Press, New York, 1992).
- PEGs are soluble in most solvents, including water, and are highly hydrated in aqueous environments, with two or three water molecules bound to each ethylene glycol segment; this has the effect of preventing adsorption either of other polymers or of proteins onto PEG-modified surfaces.
- PEGs are known to be nontoxic and not to harm active proteins or cells, whilst covalently linked PEGs are known to be non-immunogenic and non-antigenic.
- PEGs may readily be modified and bound to other molecules with only little effect on their chemistry. Their advantageous solubility and biological properties are apparent from the many possible uses of PEGs and copolymers thereof, including block copolymers such as PEG-polyurethanes and PEG-polypropylenes.
- Appropriate molecular weights for PEG spacers used in accordance with the invention may, for example, be between 120 Daltons and 20 kDaltons.
- the major mechanism for uptake of particles by the cells of the reticuloendothelial system (RES) is opsonisation by plasma proteins in blood; these mark foreign particles which are then taken up by the RES.
- the biological properties of PEG spacer elements used in accordance with the invention may serve to increase contrast agent circulation time in a similar manner to that observed for PEGylated liposomes (see e.g. Klibanov, A. L. et al. in FEBS Letters (1990) 268, 235-237 and Blume, G. and Cevc, G. in Biochim. Biophys. Acta (1990) 1029, 91-97).
- vectors include partial or complete deglycosidation by neuraminidase, endoglycosydases or periodate, since deglycosidation often results in less uptake by liver, spleen, macrophages etc., whereas neo-glycosylation of proteins often results in increased uptake by the liver and macrophages); preparation of truncated forms by proteolytic cleavage, leading to reduced size and shorter half life in circulation; and cationisation, e.g. as described by Kumagi et al. in J. Biol. Chem . (1987) 262, 15214-15219; Triguero et al. in Proc. Natl.
- Increased coupling efficiency to areas of interest may also be achieved using antibodies bound to the terminii of PEG spacers (see e.g. Maruyama, K. et al. in Biochim. Biophys. Acta (1995) 1234, 74-80 and Hansen, C. B. et al. in Biochim. Biophys. Acta (1995) 1239, 133-144).
- a PEG component as a stabiliser in conjunction with a vector or vectors or directly to the reporter in the same molecule where the PEG does not serve as a spacer.
- Other representative spacer elements include structural-type polysaccharides such as polygalacturonic acid, glycosaminoglycans, heparinoids, cellulose and marine polysaccharides such as alginates, chitosans and carrageenans; storage-type polysaccharides such as starch, glycogen, dextran and aminodextrans; polyamino acids and methyl and ethyl esters thereof, as in homo- and co-polymers of lysine, glutamic acid and aspartic acid; and polypeptides, oligonucleotides and oligosaccharides, which may or may not contain enzyme cleavage sites.
- structural-type polysaccharides such as polygalacturonic acid, glycosaminoglycans, heparinoids, cellulose and marine polysaccharides such as alginates, chitosans and carrageenans
- storage-type polysaccharides such as starch, glycogen, dextran and aminod
- spacer elements may contain cleavable groups such as vicinal glycol, azo, sulfone, ester, thioester or disulphide groups.
- Poly[N-(2-hydroxyethyl)methacrylamides] are potentially useful spacer materials by virtue of their low degree of interaction with cells and tissues (see e.g. Volfová, I., R ⁇ hová, B. and V. R. and Vetvicka, P. in J. Bioact. Comp. Polymers (1992) 7, 175-190).
- Work on a similar polymer consisting mainly of the closely related 2-hydroxypropyl derivative showed that it was endocytosed by the mononuclear phagocyte system only to a rather low extent (see Goddard, P., Williamson, I., Bron, J., Hutchkinson, L. E., Nicholls, J. and Petrak, K. in J. Bioct. Compat. Polym . (1991) 6, 4-24. ).
- Linking agents used in accordance with the invention will in general bring about linking of vector to reporter or reporter to reporter with some degree of specificity, and may also be used to attach one or more therapeutically active agents.
- Ultrasound imaging modalities which may be used in accordance with the invention include two- and three-dimensional imaging techniques such as B-mode imaging (for example using the time-varying amplitude of the signal envelope generated from the fundamental frequency of the emitted ultrasound pulse, from sub-harmonics or higher harmonics thereof or from sum or difference frequencies derived from the emitted pulse and such harmonics, images generated from the fundamental frequency or the second harmonic thereof being preferred), colour Doppler imaging and Doppler amplitude imaging, and combinations of the two latter with any of the modalities (techniques) above.
- B-mode imaging for example using the time-varying amplitude of the signal envelope generated from the fundamental frequency of the emitted ultrasound pulse, from sub-harmonics or higher harmonics thereof or from sum or difference frequencies derived from the emitted pulse and such harmonics, images generated from the fundamental frequency or the second harmonic thereof being preferred
- colour Doppler imaging and Doppler amplitude imaging and combinations of the two latter with any of the modalities (techniques) above.
- successive images of tissues such as the heart or kidney may be collected with the aid of suitable synchronisation techniques (e.g. gating to the ECG or respiratory movement of the subject).
- suitable synchronisation techniques e.g. gating to the ECG or respiratory movement of the subject.
- Measurement of changes in resonance frequency or frequency absorption which accompany arrested or retarded microbubbles may also usefully be made to detect the contrast agent.
- the present invention provides a tool for therapeutic drug delivery in combination with vector-mediated direction of the product to the desired site.
- therapeutic or “drug” is meant an agent having a beneficial effect on a specific disease in a living human or non-human animal.
- combinations of drugs and ultrasound contrast agents have been proposed in, for example, WO-A-9428873 and WO-A-9507072, these products lack vectors having affinity for particular sites and thereby show comparitively poor specific retention at desired sites prior to or during drug release.
- Therapeutic compounds used in accordance with the present invention may be encapsulated in the interior of the microbubbles or attached to or incorporated in the encapsulating walls.
- the therapeutic compound may be linked to a part of the wall, for example through covalent or ionic bonds, or may be physically mixed into the encapsulating material, particularly if the drug has similar polarity or solubility to the membrane material, so as to prevent it from leaking out of the product before it is intended to act in the body.
- the release of the drug may be initiated merely by wetting contact with blood following administration or as a consequence of other internal or external influences, e.g. dissolution processes catalyzed by enzymes or the use of of ultrasound.
- the destruction of gas-containing microparticles using external ultrasound is a well known phenomenon in respect of ultrasound contrast agents, e.g. as described in WO-A-9325241; the rate of release may be varied depending on the type of therapeutic application, using a specific amount of ultrasound energy from the transducer.
- the therapeutic agent may be covalently linked to the encapsulating membrane surface using a suitable linking agent, e.g. as described herein.
- a suitable linking agent e.g. as described herein.
- lipidated drug molecules which do not require processing to liberate an active drug are incorporated directly into the membrane.
- the active lipidated-drug can be released by increasing the strength of the ultrasound beam.
- Exemplary drug delivery systems suitable for use in the present compositions include any known therapeutic drugs or active analogues thereof containing thiol groups which are coupled to thiol containing microbubbles under oxidative conditions yielding disulphide bridges.
- the drug/vector modified microbubbles are allowed to accumulate in the target tissue.
- Administration of a reducing agent such as reduced glutathione then liberates the drug molecule from the targeted microbubble in the vicinity of the target cell increasing the local concentration of the drug and enhancing therapeutic effect.
- the product may also be prepared without the therapeutic if desired.
- the drug may then be coupled to or coated on the microbubbles prior to use.
- a therapeutic could be added to a suspension of microbubbles in aqueous media and shaken in order to attach or adhere the therapeutic to the microbubbles.
- Other drug delivery systems include vector modified phospholipid membranes doped with lipopeptide structures comprising a poly-L-lysine or poly-D-lysine chain in combination with a targeting vector.
- the microbubble carrier is condensed with DNA or RNA via elecrostatic interaction with the polycation. This method has the advantage that the vector or vectors used for targeted delivery are not directly attached to the polysine carrier moiety.
- the polylysine chain is also anchored more tightly in the microbubble membrane due to the presence of the lipid chains.
- the use of ultrasound to increase the effectiveness of delivery is also considered useful.
- free polylysine chains are firstly modified with drug or vector molecules then condensed onto the negative surface of targeted microbubbles.
- drugs useful in accordance with the invention include antineoplastic agents such as vincristine, vinblastine, vindesine, busulfan, chlorambucil, spiroplatin, cisplatin, carboplatin, methotrexate, adriamycin, mitomycin, bleomycin, cytosine arabinoside, arabinosyl adenine, mercaptopurine, mitotane, procarbazine, dactinomycin (antinomycin D), daunorubicin, doxorubicin hydrochloride, taxol, plicamycin, aminoglutethimide, estramustine, flutamide, leuprolide, megestrol acetate, tamoxifen, testolactone, trilostane, amsacrine (m-AMSA), asparaginase (L-asparaginase), etoposide, interferon a-2a and 2b, blood products such as antineoplastic agents
- RNA DNA of natural or synthetic origin, including recombinant RNA and DNA.
- DNA encoding certain proteins may be used in the treatment of many different types of diseases. For example, tumor necrosis factor or interleukin-2 genes may be provided to treat advanced cancers; thymidine kinase genes may be provided to treat ovarian cancer or brain tumors; interleukin-2 genes may be provided to treat neuroblastoma, malignant melanoma or kidney cancer; and interleukin-4 genes may be provided to treat cancer.
- Lipophilic derivatives of drugs linked to the microbubble wall through hydrophobic interactions may exhibit therapeutic effects as part of the microbubble or after release from the microbubble, e.g. by use of ultrasound.
- a lipophilic group may be introduced for anchoring the drug to the membrane.
- the lipophilic group should be introduced in a way that does not influence the in vivo potency of the molecule, or the lipophilic group may be cleaved releasing the active drug.
- Lipophilic groups may be introduced by various chemical means depending on functional groups available in the drug molecule. Covalent coupling may be effected using functional groups in the drug molecule capable of reacting with appropriately functionalised lipophilic compounds.
- lipophilic moieties include branched and unbranched alkyl chains, cyclic compounds, aromatic residues and fused aromatic and non-aromatic cyclic systems. In some instances the lipophilic moiety will consist of a suitably functionalised steroid, like cholesterol and related compounds.
- functional groups particularly suitable for derivatisation include nucleophilic groups like amino, hydroxy and sulfhydryl groups.
- Suitable processes for lipophilic derivatisation of any drug containing a sulfhydryl group, like captopril may include direct alkylation, e.g. reaction with an alkyl halide under basic conditions and thiol ester formation by reaction with an activated carboxylic acid.
- derivatisation of any drug having carboxylic functions like atenolol and chlorambucil, include amide and ester formation by coupling of amines and alcohols, respectively, possesing requested physical properties.
- a preferred aspect is attachment of cholesterol to a therapeutic compound by forming a degradable ester bond.
- a preferred application of the present invention relates to angiogenesis, which is the formation of new blood vessels by branching from existing vessels.
- the primary stimulus for this process may be inadequate supply of nutrients and oxygen (hypoxia) to cells in a tissue.
- the cells may respond by secreting angiogenetic factors, of which there are many; one example is vascular endothelial growth factor. These factors initiate the secretion of proteolytic enzymes which break down the proteins of the basement membrane, as well as inhibitors which limit the action of these potentially harmful enzymes.
- the combined effect of loss of attachment and signals from the receptors for angiogenetic factors is to cause the endothelial cells to move, multiply, and rearrange themselves, and finally to synthetise a basement membrane around the new vessels.
- Tumors must initiate angiogenesis when they reach millimeter size in order to keep up their rate of growth.
- angiogenesis is accompanied by characteristic changes in the endothelial cells and their environment, this process is a promising target for therapeutic intervention.
- the transformations accompanying angiogenesis are also very promising for diagnosis, a preferred example being malignant disease, but the concept also shows great promise in inflammation and a variety of inflammation-related diseases. These factors are also involved in re-vascularisation of infarcted parts of the myocardium, which occurs if a stenosis is released within a short time.
- angiogenesis mav be detected by the majority of the imaging modalities in use in medicine. Contrast-enhanced ultrasound may possess additional advantages, the contrast medium being microspheres which are restricted to the interior of blood vessels. Even if the target antigens are found on many cell types, the microspheres will attach exclusively to endothelial cells.
- prodrugs may also be used in agents according to the invention.
- drugs may be derivatised to alter their physicochemical properties and to adapt them for inclusion into the reporter; such derivatised drugs may be regarded as prodrugs and are usually inactive until cleavage of the derivatising group regenerates the active form of the drug.
- Another alternative is to incorporate the prodrug, the prodrug-activating enzyme and the vector in the same microbubble in a system where the prodrug will only be activated after some external stimulus.
- a stimulus may, for example, be a tumour-specific protease as described above, or bursting of the bubbles by external ultrasound after the desired targeting has been achieved.
- Therapeutics may easily be delivered in accordance with the invention to diseased or necrotic areas including the heart and vasculature in general, and to the liver, spleen and kidneys and other regions such as the lymph system, body cavities or gastrointestinal system.
- Products according to the present invention may be used for targeted therapeutic delivery either in vivo or in vitro.
- the products may be useful in in vitro systems such as kits for diagnosis of different diseases or characterisation of different components in blood or tissue samples.
- Similar techniques to those used to attach certain blood components or cells to polymer particles(e.g. monodisperse magnetic particles) in vitro to separate them from a sample may be used in the present invention,using the low density of the reporter units in agents of the present invention to effect separation of the gas-containing material by floatation and repeated washing.
- Vectors which may be usefully employed in generating multiple-specific targetable contrast agents according to the invention include the following:
- peptide vectors and lipopeptides thereof of particular interest for targeted ultrasound imaging are listed below: Atherosclerotic plaque binding peptides such as YRALVDTLK, YAKFRETLEDTRDRMY and RALVDTEFKVKQEAGAK; Thrombus binding peptides such as NDGDFEEIPEEYLQ and GPRG; Platelet binding peptides such as PLYKKIIKKLLES; and cholecystokinin, ⁇ -melanocyte-stimulating hormone, heat stable enterotoxin 1, vasoactive intestinal peptide, synthetic alpha-M2 peptide from the third heavy chain complementarity-determining region and analogues thereof for tumor targeting.
- Atherosclerotic plaque binding peptides such as YRALVDTLK, YAKFRETLEDTRDRMY and RALVDTEFKVKQEAGAK
- Thrombus binding peptides such as NDGDFEEIPEEYLQ and GPRG
- Protein and Peptide Vectors Antibodies Vector type Receptor Comments/areas of use Ref antibodies CD34 vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies ICAM-1 vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies ICAM-2 vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies ICAM-3 vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies E-selectin vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies P-selectin vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies PECAM vascular diseases in general, (general) normal vessel wall (
- VLA-1 normal vessel wall (e.g VLA-2, VLA-3, myocardium), activated VLA-4, VLA-5, endothelium, immune cells VLA-6, ⁇ 1 ⁇ 7 , ⁇ 1 ⁇ 8 , ⁇ 1 ⁇ V , LFA-1, Mac-1, CD41a, etc.
- Vector type Receptor Comments/areas of use Ref L-selectin CD34 vascular diseases in MadCAM1 general, normal vessel wall GlyCam 1 (e.g myocardium), activated endothelium, Lymph nodes Other selectins carbohydrate vascular diseases in ligands general, normal vessel wall (sialyl Lewis x) (e.g myocardium), activated heparan sulfate endothelium RGD-peptides integrins angiogenisisis PECAM PECAM, Endothelium, and other Cells in immune system Integrins, Laminin, Endothelium, e.g.
- Integrin Integrins, Cells in immune system receptors e.g. VLA-1, vessel wall e.g. Laminin, VLA-2, VLA-3, etc.
- Vectors Comprising Cytokines/Growth Factors/Peptide Hormones and Fragments Thereof Vector type Receptor Comments/areas of use Ref Epidermal growth EGF-receptor or Tumours factor related receptors Nerve growth NGF-receptor Tumours factor Somatostatin ST-receptor Tumours Endothelin Endothelin- Vessel wall receptor Interleukin-1 IL-1-receptor Inflammation, activated cells of different kinds Interleukin-2 IL-2-receptor Inflammation, activated cells of different kinds Chemokines (ca.
- apolipoprotein B receptors e.g., LDL receptor
- Apolipoprotein E LDL receptor proteoglycans Adhesion- proteoglycans promoting proteins, e.g. Purpurin Viral coat proteoglycans proteins, e.g. HIV, Herpes Microbial “Antigen 85” fibronectin, collagen, adhesins complex of fibrinogen, vitronectin, mycobacteria heparan sulfate ⁇ -amyloid proteoglycans ⁇ -amyloid accumulates in precursor Alzheimer's disease Tenascin, heparan sulfate, e.g. tenascin C integrins
- Vectors Comprising Non-peptide Agonists/Antagonists of Cytokines/Growth Factors/Peptide Hormones/Cell Adhesion Molecules Vector type Receptor Comments/areas of use Ref Endothelin Endothelin Vessel wall antagonist receptor Desmopressin Vasopressin Kidney (vasopressin receptor Vessel wall analogue) Demoxytocin Oxytocin Reproductive organs, (oxytocin Receptor Mammary glands, analogue) Brain Angiotensin II Angiotensin II Vessel wall receptor receptors brain antagonists adrenal gland CV-11974, TCV-116 non-peptide RGD- integrins Cells in immune system analogues vessel wall etc.
- Vectors Comprising Anti-angiogenic Factors Vector type Target Comments/areas of use Ref Angiostatin EC of tumors plasminogen fragment K cartilage-derived EC of tumors J inhibitor ⁇ -Cyclodextrin tumors, C tetradecasulfate inflammation fumagillin and analogs tumors, E inflammation Interferon- ⁇ EC of tumors K Interferon- ⁇ EC of tumors E interleukin-12 EC of tumors E linomide tumors, A inflammation medroxyprogesterone EC of tumors K metalloproteinase EC of tumors K inhibitors pentosan polysulfate EC of tumors K platelet factor 4 EC of tumors M Somatostatin EC of tumors K Suramin EC of tumors K Taxol EC of tumors K thalidomide EC of tumors K Thrombospondin EC of tumors K
- Vectors Comprising Angiogenic Factors Comments/areas Vector type Target of use Ref acidic fibroblast EC of tumors K growth factor adenosine EC of tumors K Angiogenin EC of tumors K Angiotensin II EC of tumors K basement membrane tumors e.g., tenascin, M components collagen IV basic fibroblast EC of tumors K growth factor Bradykinin EC of tumors K Calcitonin gene- EC of tumors K related peptide epidermal growth EC of tumors K factor Fibrin tumors K Fibrinogen tumors K Heparin EC of tumors K histamine EC of tumors K hyaluronic acid EC of tumors K or fragments thereof Interleukin-1 ⁇ EC of tumors K laminin, laminin EC of tumors K fragments nicotinamide EC of tumors K platelet acti- EC of tumors K vating factor Platelet-derived EC of tumors K endot
- Ref angiopoietin tumors B inflammation ⁇ 2 -antiplasmin tumors, inflammation combinatorial tumors, for instance: compounds libraries, compounds inflammation that bind to basement from membrane after degradation endoglin tumors, D inflammation endosialin tumors, D inflammation endostatin [collagen tumors, M fragment] inflammation Factor VII related tumors, D antigen inflammation fibrinopeptides tumors, ZC inflammation fibroblast growth tumors, E factor, basic inflammation hepatocyte growth tumors, I factor inflammation insulin-like growth tumors, R factor inflammation interleukins tumors, e.g.,: IL-8 I inflammation leukemia inhibitory tumors, A factor inflammation metalloproteinase tumors, e.g., batimastat E inhibitors inflammation Monoclonal antibodies tumors, for instance: to inflammation angiogen
- Receptors/Targets Associated with Angiogenesis Vector type Target Comments/areas of use Ref biglycan tumors, dermatan sulfate X inflammation proteoglycan CD34 tumors, L inflammation CD44 tumors, F inflammation collagen type I, IV, tumors, A VI, VIII inflammation decorin tumors, dermatan sulfate Y inflammation proteoglycan dermatan sulfate tumors, X proteoglycans inflammation endothelin tumors, G inflammation endothelin receptors tumors, G inflammation fibronectin tumors P Flk-1/KDR, Flt-4 tumors, VEGF receptor D inflammation FLT-1 (fms-like tumors, VEGF-A receptor O tyrosine kinase) inflammation heparan sulfate tumors, P inflammation hepatocyte growth tumors, I factor receptor (c-met) inflammation insulin-like growth tumors, R factor/mannose-6- inflammation phosphate receptor integrins: Tumors
- Intercellular adhesion tumors P molecule-1 and -2 inflammation Jagged gene product tumors, T inflammation Ly-6 tumors, a lymphocyte activation N inflammation protein matrix tumors, D metalloproteinases inflammation MHC class II tumors, inflammation Notch gene product tumors, T inflammation Osteopontin tumors Z PECAM tumors, alias CD31 P inflammation plasminogen activator tumors, ZC receptor inflammation platelet-derived growth tumors, E factor receptors inflammation Selectins: E-, P- tumors, D inflammation Sialyl Lewis-X tumors, blood group antigen M inflammation stress proteins: tumors, molecular chaperones glucose regulated, heat inflammation shock families and others syndecan tumors, T inflammation thrombospondin tumors, M inflammation TIE receptors tumors, tyrosine kinases with Ig- E inflammation and EGF-Iike domains tissue factor tumors, Z inflammation tissue inhibitor of tumors, e.g., TIMP-2 U metalloproteinases inflammation transforming growth tumors, E factor
- Oligonucleotide Vectors Vector type Receptor Comments/areas of use Ref Oligonucleotides DNA made Tumours complementary to available by Myocardial infarction repeated necrosis All other diseases that sequences, e.g. involves necrosis genes for ribosomal RNA, Alu-sequences Oligonucleotides DNA made Tumours complementary to available by disease-specific necrosis in a mutations (e.g. region of the mutated relevant disease oncogenes). Oligonucleotides DNA of infective Viral or bacterial complementary to agent infections DNA of infecting agent.
- Modified Oligonucleotide Vectors Vector type Receptor Comments/areas of use Ref Phosphorothioate As for As for unmodified oligos oligos unmodified oligos 2′-O-methyl As for As for unmodified oligos substituted unmodified oligos oligos circular oligos As for As for unmodified oligos unmodified oligos oligos As for As for unmodified oligos containing unmodified hairpin oligos structure to decrease degradation oligos with As for As for unmodified oligos terminal unmodified phosphorothioate oligos 2′-fluoro oligos As for As for unmodified oligos unmodified oligos 2′-amino oligos As for As for unmodified oligos unmodified oligos DNA-binding As for Increased binding affinity drugs conjugated unmodified as compared to pure oligos to oligos (
- Nucleoside and Nucleotide Vectors Vector type Receptor Comments/areas of use Ref Adenosine or Adenosine Vessel wall analogues receptors Heart ADP, UDP, UTP Various Many tissues, e.g. brain, and others nucleotide spinal cord, kidney, spleen receptors
- Receptors Comprising DNA-binding Drugs
- Vector type Receptor Comments/areas of use Ref acridine DNA made Tumours, derivatives available by Myocardial infarction and distamycin necrosis all other diseases involving netropsin necrosis or other processes actinomycin D liberating DNA from cells echinomycin bleomycin etc.
- Receptors Comprising Protease Substrates
- Vector type Receptor Comments/areas of use Ref Peptidic or non- Cathepsin B Tumours, a variety of which peptidic may more or less specifically substrates overexpress proteases of various kinds, e.g. Cathepsin B
- Receptors Comprising Protease Inhibitors
- Vector type Receptor Comments/areas of use Ref Peptidic or non- Cathepsin B Tumours, a variety of which peptidic may more or less specifically inhibitors overexpress proteases of e.g. N-acetyl- various kinds, e.g. Leu-Leu- Cathepsin B norleucinal bestatin Aminopeptidases Tumours, ([(2S,3R)-3- e.g.
- drugs useful in accordance with the invention include: abamectin, abundiazole, acaprazine, acabrose, acebrochol, aceburic acid, acebutolol, acecainide, acecarbromal, aceclidine, ac clof nac, acedapsone, acediasulfone, acedoben, acefluranol, acefurtiamine, acefylline clofibrol, acefylline piperazine, aceglatone, aceglutamide, aceglutamide aluminium, acemetacin, acenocoumarol, aceperone, acepromazine, aceprometazine, acequinoline, acesulfame, acetaminophen, acetaminosalol, acetanilide, acetarsone, acetazolamide, acetergamine, acetiamine
- targeted agents to bind specifically to cells expressing a target may be studied by microscopy and/or using a flow chamber containing immobilised cells, for example employing a population of cells expressing the target structure and a further population of cells not expressing the target.
- Radioactive, fluorescent or enzyme-labelled streptavidin/avidin may be used to analyse biotin attachment.
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Ile-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- DSPS vanti, 4.5 mg
- lipopeptide from a 0.5 mg
- DSPS 0.8 mL
- a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial.
- the mixture was warmed to 80° C. for 5 minutes (vials shaken during warming).
- the samples were cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles rolled overnight.
- Bubbles were washed several times with deionised water and analysed by Coulter counter (Size: 1-3 micron (87%), 3-5 micron (11.5%)) and acoustic attenuation (frequency max att.: 3.5 MHz).
- the microbubbles were stable at 120 mm Hg.
- the human endothelial cell line ECV 304 derived from a normal umbilical cord (ATCC CRL-1998) was cultured in 260 mL Nunc culture flasks (chutney 153732) in RPMI 1640 medium (Bio Whittaker) to which L-Glutamine 200 mM, Penicillin/Streptomycin (10.000 U/mL and 10.000 mcg/mL) and 10% Fetal Bovine Serum (Hyclone Lot no. AFE 5183) were added.
- Cover-glasses, 22mm in diameter (BDH, Cat no. 406/0189/40) were sterilised and placed on the bottom of 12 well culture plates (Costar) before cells in 0,5 mL complete medium with serum was added on top.
- the chamber consists of a groove carved into a glass plate upon which the cover slip with cells was placed with the cells facing the groove forming a flow channel.
- Ultrasound microbubbles from section b) were passed from a reservoir held at 37 degree Celsius through the flow chamber and back to the reservoir using a peristaltic pump. The flow rate was adjusted to simulate physiological relevant shear rates.
- the flow chamber was placed under a microscope and the interaction between the microspheres and cells viewed directly. A camera mounted on the microscope was connected to a colour video printer and a monitor.
- a 22 kg mongrel dog was anaesthetized with pentobarbital and mechanically ventilated.
- the chest was opened by a midline sternotomy, the anterior pericardium was removed, and a 30 mm gelled silicone rubber spacer was inserted between the heart and a P5-3 transducer of an ATL HDI-3000 ultrasound scanner.
- the scanner was set for intermittent short axis imaging once in each end-systole by delayed EGC triggering.
- a net volume of 2 mL of microbubbles from b) were injected as a rapid intravenous bolus. 3 seconds later, the imaged right ventricle was seen to contain contrast material, another 3 seconds later, the left ventricle was also filled, and a transient attenuation shadow that obscured the view of the posterior parts of the left ventricle was observed. A substantial increase in brightness of the myocardium was seen, also in the portions of the heart distal to the left ventricle when the attenuation shadow subsided.
- the ultrasound scanner was set to continuous, high frame rate high output power imaging, a procedure known to cause destruction of utrasound contrast agent bubbles in the imaged tissue regions. After a few seconds, the scanner was adjusted back to its initial setting. The myocardium was then darker, and closer to the baseline value. Moving the imaged slice to a new position resulted in re-appearance of contrast effects, moving the slice back to the initial position again resulted in a tissue brightness again close to baseline.
- a net volume of 2 mL microbubbles prepared in an identical manner to b) above with the exception that no lipopeptide was included in the preparation was injected, using the same imaging procedure as above.
- the myocardial echo enhancement was far less intense and of shorter duration than observed in case 1.
- At the completion of the left ventricular attenuation phase there was also almost complete loss of myocardial contrast effects, and a myocardial echo increases in the posterior part of the left ventricle as in case 1 was not observed.
- This example is directed at the preparation of targeted microbubbles comprising multiple peptidic vectors.
- DSPE distearoyl phosphatidyl ethanolamine
- the RGDC peptide was synthesised on a ABI 433A automated peptide synthesiser (0.25 mmol scale, Fmoc-Cys(Trt)-Wang resin, (Novabiochem). All amino acids were activated using HBTU.
- the crude peptide was removed from the resin and simultaneously deprotected in TFA containing 5% EDT, 5% phenol and 5% water. Following evaporation of the excess cleavage solution the peptide was precipitated and triturated several times with diethyl ether before air drying.
- the crude peptide was purified by preparative hplc and fractions containing pure product combined and freeze dried. Final characterisation was performed using analytical hplc and MALDI MS.
- DSPS (Avanti, 5.0 mg), lipopeptide (0.5 mg) from example 1 a) and PE-PEG-MAL (0.5 mg) from section a) was weighed into a clean vial and 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was sonicated for 3-5 mins, warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter. The mixture was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes.
- the infranatant was exchanged with 1 mL of PBS containing 1 mg of the peptide RGDC and the pH adjusted to 8. The conjugation reaction was allowed to proceed for 2 h. The bubbles were washed in PBS then with water until all unreacted RGDC had been removed from the infranatant as observed by MALDI-MS. The microbubbles were further analysed by Coulter counter (98% between 1 and 7 micron).
- This example is directed at the preparation of microbubbles comprising multiple antibody vectors for targeted ultrasound.
- DSPS Advanti, 4.5 mg
- PE-PEG 2000 -Maleimide from example 2 a 0.5 mg
- the mixture was warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter.
- the sample was cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles washed three times with distilled water.
- the PEG spacer length may also be varied to include longer e.g. PEG 3400 and PEG 5000 or shorter e.g. PEG 600 or PEG 800 chains. Addition of a third antibody such as thiolated-anti-CD34 is also envisaged. EXAMPLE 4
- the peptide was synthesised on an ABI 433A automatic peptide synthesiser starting with Fmoc-Ile-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids were preactivated using HBTU before coupling.
- DSPS (5 mg, Avanti) was weighed into a clean vial along with poly-L-lysine (Sigma, 0.2 mg) and peptide from a) above (0.2 mg). To the vial was added 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol. The mixture was warmed to 80° C. for 5 minutes. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes.
- the spacer element contained within the PS binding/Fibronectin fusion peptide can also be replaced with other spacers such as PEG 2000 or poly alanine (-AAA-). It is also envisaged that a form of pre-targeting may be employed, whereby the DSPS binding/Fibronectin fragment fusion peptide is firstly allowed to associate with cells via the fibronectin peptide binding. This is followed by administration of PS microbubbles which then bind to the PS binding peptide.
- This example is directed at the preparation of targeted ultrasound microbubbles whereby streptavidin is used as a linker between biotinylated reporter(s) and vector(s).
- the peptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Trp(Boc)-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids were preactivated using HBTU before coupling.
- This peptide was synthesised and purified using similar protocols to those described in section b) above.
- the pure product was characterised by hplc and MALDI MS.
- DSPS (Avanti, 4.5 mg) and biotin-PEG 3400 - ⁇ -Alanine cholesterol from section a) (0.5 mg) were weighed into a vial and 0.8 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming). The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vial was shaken in a cap mixer for 45 s and the microbubbles rolled overnight.
- microbubble suspension was washed several times with deionised water and analysed by Coulter counter and acoustic attenuation.
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Gly-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- DSPS vanti, 4.5 mg
- lipopeptide from a 0.5 mg
- DSPS 0.8 mL
- a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial.
- the mixture was warmed to 80° C. for 5 minutes (vials shaken during warming).
- the samples were cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles formed rolled overnight.
- the microbubbles were washed several times with deionised water and analysed by Coulter counter and acoustic attenuation.
- microbubble preparation from b) above was treated in an analogous manner to that described in example 5 section e).
- NH 2 -PEG 3400 -DSPE is carboxylated using succinic anhydride, e.g. by a similar method to that described by Nayar, R. and Schroit, A. J. in Biochemistry (1985) 24, 5967-71.
- Streptavidin is covalently bound to Succ-PEG 3400 -DSPE in the membrane by standard coupling methods using a water-soluble carbodiimide.
- the sample is placed on a roller table during the reaction. After centrifugation the infranatant is exchanged with water and the washing is repeated.
- the functionality of the attached streptavidin is analysed by binding, e.g. to fluorescently labeled biotin, biotinylated antibodies (detected with a fluorescently labeled secondary antibody) or biotinylated and fluorescence- or radioactively-labeled oligonucleotides. Analysis is performed by fluorescence microscopy or scintillation counting.
- Microbubbles from section c) are incubated in a solution containing human transferrin and human endothelium IgG antibody biotinylated using the method described by Bayer et al., Meth. Enzymol., 62, 308.
- the vector-coated microbubbles are washed as described above.
- Microbubbles from section c) are incubated in a solution containing a mixture of biotin-GAAAGGTAGTGGGGTCGTGTGCCGG and biotin-GGCGCTGATGATGTTGTTGATTCTT.
- the oligonucleotide-coated microbubbles are washed as described above. Binding of the oligonucleotide to the bubbles is detected e.g. by using fluorescent-labeled oligonucleotides for attachment to the bubbles, or by hybridising the attached oligonucleotide to a labeled (fluorescence or radioactivity) complementary oligonucleotide.
- the functionality of the oligonucleotide-carrying microbubbles is analysed, e.g. by hybridising the bubbles with immobilized DNA-containing sequences complementary to the attached oligonucleotide.
- oligonucleotide complementary to ribosomal DNA of which there are many copies per haploid genome
- an oligonucleotide complementary to an oncogene e.g. ras of which there is one copy per haploid genome
- DSPS Advanti, 4.5 mg
- DSPE Advanti, 1.0 mg
- the mixture was warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter.
- the sample was cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vial was shaken in a cap mixer for 45 s and the microbubbles washed two times with distilled water then resuspended in 0.1 M sodium borate buffer pH 9.
- Fibronectin 0.5 mg
- transferrin 1.3 mg
- PBS PBS
- a solution containing NHS-fluorescin in DMSO added.
- the mixture was stirred at room temperature for 1 hour then the protein purified on a Superdex 200 column.
- the fluorescein-labelled protein mixture in phosphate buffer pH 7.5 was freeze dried.
- the freeze-dried product from b) was re-dissolved in 0.5 mL water and to the fluorescein labelled fibronectin/transferrin mixture was added 0.1 mmol of the crosslinker SDBP (Pierce).
- the solution was incubated on ice for 2 hours, charged on a NAP-5 column and eluted with PBS.
- This example is directed at the preparation of polymeric ultrasound contrast agents comprising multiple vectors attached to non-surfactant for targeting/therapeutic applications.
- Hollow polymer particles of P73 (as described in patent WO 96/07434) containing avidin were prepared by a process involving the freeze-drying of an oil-in-water emulsion using the following procedure: An oil solution was prepared by dissolving 0.25 g of the biodegradable polymer P73 [poly(ethylidene bis(16-hydroxyhexadecanoate)co(adipic acid)] in 5 mL of camphene at 60° C. To 0.2 mL of the oil solution was added 2 mg avidin.
- aqueous solution was then prepared by dissolving 0.4 g of the polymer, a-(16-hexadecanoyloxyhexadecanoyl)-w-methoxypolyoxyethylene ester, in 20 mL of water at 60° C.
- the oil solution (0.2 mL) was then mixed with of the aqueous solution (0.8 mL) in a vibromixer (Capmix) for 15 s to form the oil-in-water emulsion.
- the emulsion was frozen in dry ice and methanol then dried at a pressure of 200 mTorr for 24 h to remove excess solvent.
- the powder was reconstituted as a suspension of hollow particles by addition of 1.0 mL water.
- the resulting ultrasound contrast agent was confirmed by microscopy observation, Coulter size distribution, acoustic attenuation and resistance to external pressure.
- the particles from a) were centrifuged and the supernatant replaced with 1 mL of PBS buffer pH 7.5 containing 0.2 mg of biotin-GGCGCTGATGATGTTGTTGATTCTT and 0.2 mg of biotin-D-Trp-Leu-Asp-Ile-Ile-Trp.OH from b) above. After incubation for 24 h the particles were washed extensively with PBS and water.
- a homogeneous suspension of GAM (6 ⁇ 10 8 particles/mL) in 5 mg/mL albumin was used, with all manipulations being carried out at room temperature.
- Two 10 mL aliquots were centrifuged (170 ⁇ g, 5 minutes) to promote floatation of the microspheres and 8 mL of the underlying infranatant was removed by careful suction and replaced by an equal volume of air-saturated phosphate buffered saline, the preparations being rotated for 15-20 minutes to resuspend the microspheres. This procedure was repeated twice, whereafter only negligible amounts of free non-microsphere-associated albumin were assumed to remain.
- streptavidin conjugated to horseradish peroxidase was added to both suspensions and the tubes were rotated for 1 hour to allow for reaction.
- the microspheres were then washed three times, resuspended in 100 mM citrate-phosphate buffer (pH 5) containing 0.1 mg/mL phenylenediamine dihydrochloride and 0.01% hydrogen peroxide, and rotated for 10 minutes. Development of a yellow-green colour was indicative of the presence of enzyme.
- biotinylated microspheres are then used to prepare multiple-specific targeting products in an analogous manner to those exemplified in examples 5), 6) and 7).
- the lipopeptide was synthesised as described in example 1) using commercially available amino acids and polymers.
- DSPS Advanti, 4 mg and lipopeptide from a) (0.5 mg, 0.2 mmol) and lipopeptide from b) (0.5 mg) were weighed into each of 2 vials and 0.8 mL of a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming). The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles formed rolled overnight.
- microbubbles were washed several times with deionised water and analysed by MALDI mass spectrometry as described in example 1 b).
- the microbubbles following analysis by microscopy were seen to consist of a range of sizes between 1 and 5 micron. Furthermore the microbubbles were fluorescent.
- This example is directed at the preparation of multiple vector targeted ultrasound agents.
- the lipopeptide shown below was synthesised on a ABI 433A automatic peptide synthesiser starting with a Rink amide resin on a 0.1 mmol scale using 1 mmol amino acid cartridges.
- DSPS vanti, 4.5 mg
- lipopeptide from a) 0.5 mg
- PE-PEG 2000 -Maleimide from example 2 0.5 mg
- the mixture was warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter.
- the sample was cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles washed three times with distilled water.
- FITC labelled CD71 anti-transferrin receptor Ab 100 mg/mL, Becton Dickinson
- 0.7 mL in PBS was modified with Traut's reagent (0.9 mg, Pierce) at room temperature for 1 h.
- Excess reagent was separated from modified protein on a NAP-5 column (Pharmacia).
- the lipid structure shown above was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Cys(Trt)-Wang resin (Novabiochem) on a 0.25 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU coupling chemistry.
- DSPS vanti, 4.5 mg
- the lipid structure from a) above 0.5 mg
- the mixture was warmed to 80° C. for 5 minutes (vials shaken during warming) and filtered while still hot through a 40 micron filter.
- the samples were cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles placed on roller table overnight. Bubbles were washed several times with deionised water and analysed for thiol group incorporation using Ellmans Reagent.
- This example is directed to the preparation of microbubbles having a reactive group on the surface for non-specific targeting, principally utilising disulphide exchange reactions to effect binding to a multiplicity of cellular targets.
- DSPS (Avanti, 5.0 mg) and the thiol containing lipid structure from example 15 a)(1.0 mg) were weighed into a clean vial and 0.8 mL of a solution containing 1.4% propylene glycol/2.4% glycerol in water added. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming) and filtered while still hot through a 40 micron filter. The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles placed on roller table overnight. Bubbles were washed several times with deionised water and analysed for thiol group incorporation using Ellmans Reagent.
- the structure shown above was synthesised using a manual nitrogen bubbler apparatus starting with Fmoc protected Rink Amide MBHA resin (Novabiochem) on a 0.125 mmol scale. All amino acids were purchased from Novabiochem and palmitic acid from Fluka. Coupling was carried out using standard TBTU/HOBt/DIEA protocols. Bromoacetic acid was coupled through the side-chain of Lys as a symmetrical anhydride using DIC preactivation. Captopril (Sigma) dissolved in DMF was introduced on the solid-phase using DBU as base.
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Rink amide resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- DSPS (Avanti, 4.5 mg), product from a) (0.5 mg) and product from b) (0.5 mg) were weighed into a vial and 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming). The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were firstly shaken in a cap mixer for 45 s then rolled for 1 h followed by extensive washing with deionised water. No detectable levels of starting material were found in the final wash solution as evidenced by MALDI MS.
- This example is directed at the preparation of targeted microbubbles comprising multiple peptidic vectors having a combined targeting and a therapeutic application.
- DSPS (Avanti, 5.0 mg), lipopeptide from a)(0.3 mg)and polymixin B sulphate (Sigma, 0.5 mg) were weighed into a clean vial and 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was sonicated for 3-5 mins, warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter. The mixture was cooled to room temperature and the head space flushed with perfluorobutane gas. The vial was shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes. The microbubbles were washed in water until no polymixin B sulphate or lipopeptide could be detected in the infranatant by MALDI-MS. Microscopy showed that the size distribution of the bubble population was between 1-8 micron as desired.
- This example is directed at the preparation of targeted microbubbles comprising multiple vectors for targeted/therapeutic/drug release applications.
- the lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Ala-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- the methotrexate structure was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Cys(Trt) Tentagel resin on a 0.1 mmol scale.
- the simultaneous removal of product from the resin and deprotection of protecting groups was carried out in TFA containing 5% EDT and 5% H 2 O for 2 hours giving a crude product yield of 160 mg.
- DSPS Advanti, 4.5 mg.
- thiol containing lipopeptide from example 15 a) 0.5 mg
- lipopeptide from a) (0.2 mg) above were weighed into a clean vial and 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol added.
- the mixture was sonicated for 3-5 mins, warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter.
- the mixture was cooled to room temperature and the head space flushed with perfluorobutane gas.
- the vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes following which the infranatant was discarded.
- methotrexate structure from b) above (0.5 mg) was dissolved in PBS pH 8.0. The solution was then added to the thiol containing bubbles from c) and disulphide bond formation allowed to proceed for 16 h. Following extensive washing with PBS and water the bubbles were analysed by microscopy and MALDI MS.
- the disulphide bond linking the methotrexate structure to the microbubble may be reduced in vivo liberating the free drug molecule.
- This in combination with a tumour specific vector is a drug delivery system.
- a physiologically relevant reducing agent such as glutathione may be used to bring about drug release.
- This example is directed to the preparation of microbubbles for gene therapy/anti-sense applications. It is envisaged that specific targeting may be achieved by further doping of microbubble membranes with vector modified lipid structures as described in example 1.
- DSPS (Avanti, 4.5 mg) was weighed into a clean vial. 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol was added and the mixture sonicated for 2 min then warmed to 80° C. for 5 minutes. Immediately following warming the solution was filtered through a 4 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vial was shaken in a cap mixer for 45 s. Bubbles were then washed once with deionised water and the infranatant discarded. The microbubbles were then resuspended in 0.5 mL water.
- This example is directed to the preparation of microbubbles having a thiol group on the surface for modification with thiol containing vectors for targeting/drug delivery and drug release.
- the peptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Cys(Trt)-Tentagel resin on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids were preactivated using HBTU before coupling.
- microbubble bound peptide was carried out by reduction of the disulphide bond using the water souble reducing agent tris-(2-carboxyethyl)-phosphine. Following reduction the infranatant was found to contain free dabsyl-peptide as evidenced by hplc and MALDI MS.
- Gas-containing Microparticles Comprising Polymer from Ethylidene Bis(16-hydroxyhexadecanoate) and Adipoyl Chloride and Biotin-amidocaproate-Ala Covalently Attached to the Polymer
- the polymer is prepared from ethylidene bis(16-hydroxyhexadecanoate) and adipoyl chloride as described in WO-A-9607434, and a polymer fraction with molecular weight 10000 is purified using gel permeation chromatography (GPC). 10 g of the material (corresponding to 1 mmol OH groups), Z-alanine (5 mmol) and dimethylaminopyridine (4 mmol) are dissolved in dry dimethylformamide/tetrahydrofuran and dicyclohexylcarbodiimide is then added. The reaction mixture is stirred at ambient temperature overnight. Dicyclohexylurea is filtered off and the solvent is removed using rotary evaporation. The product is purified by chromatography, fractions containing the title compound are combined and the solvent is removed using rotary evaporation. The structure of the product is confirmed by NMR.
- Z-Ala-polymer (0.1 mmol) is stirred in toluene/tetrahydrofuran and glacial acetic acid (15% of the total volume) and hydrogenated in the presence of 5% palladium on charcoal for 2 hours. The reaction mixture is filtered and concentrated in vacuo.
- biotinylated microspheres are then used to prepare multiple-specific targeting products similar to those exemplified in examples 5), 6) and 7).
- Gas-containing microbubbles were prepared by mixing DSPS and biotin-PEG 3400 -acyl-phosphatidyl ethanolamine as described in example 5 a).
- microbubble suspension was divided into 0.2 mL aliquots and fluorescein conjugated streptavidin added as shown in the table below.
- the samples were incubated on a roller table for 15 or 30 minutes at ambient temperature before removal of excess protein by washing in PBS.
- biotinylated vectors or therapeutic agents may be conjugated to streptavidin or avidin coated microbubbles using this procedure.
- This example is directed at the preparation of thrombus targeted US agents comprising a therapeutic thromolytic agent.
- the lipopeptide was synthesised on a ABI 433 A automatic peptide synthesiser starting with Rink amide resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- a solution of 0.1 mL of ammonium carbonate buffer containing 0.1 mg of t-PA (Sigma) was made up to 0.2 mL by the addition of water.
- To this solution was added 0.4 mg of Sulpho-SMPB (Pierce) dissolved in 0.05 mL DMSO.
- the protein solution was left standing at room temperature for 45 min then purification carried out on a Superdex 200 column. The product was eluted in PBS and the modified protein fraction collected.
- DSPS (Avanti, 5.0 mg) was weighed into a clean vial along with 0.5 mg of the lipopeptide from a) and 0.5 mg of the thiol containing lipopeptide from example 15 a). To this was added 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol and the mixture sonicated for 2 min then warmed to 80° C. for 5 minutes. Immediately following warming the solution was filtered through a 4 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles washed 2 times with deionised water.
- the infranatant was discarded and replaced with a 1 mL aliquot of the protein solution from b) above.
- the conjugation reaction was allowed to proceed for 1 h.
- the bubbles were centrifuged and infranatant exchanged with a further 1 mL of protein solution.
- the incubation step was repeated until all protein solution was used up.
- the microbubbles were then washed extensively with water and analysed by Coulter counter. The microbubbles were tested in the flow chamber assay described in example 1 c). Microbubbles modified with protein were found to bind in higher numbers than those comprising either lipopeptide/DSPS or DSPS alone.
- a solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5.0 mg), product from a) (0.5 mg) and product from c) (0.5 mg) in a vial.
- the mixture was sonicated for 5 min and then heated at 80 ° C. for 5 min (vial was shaken during warming).
- the solution was filtered and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water.
- This example is directed at non-specific modification of a multiplicity of cell receptors on endothelial cells.
- a solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 4.5 mg) and product from a) (0.5 mg) in a vial.
- the mixture was sonicated for 5 min and then heated at 80 ° C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water.
- MALDI mass spectrometry showed no detectable level of compound from a) in the final wash solution.
- microbubbles are considered useful as targeted drug delivery agents.
- a solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5.0 mg), product from b) (0.5 mg) and c) (0.5 mg) and in a vial.
- the mixture was sonicated for 5 min and then warmed to 80° C. for 5 min (vial was shaken during warming).
- the solution was filtered and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water.
- Incorporation of atenolol containing lipopeptide and chlorabucil analogue into the bubble membrane was confirmed by MALDI-MS as described in example 1 c).
- a solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5.0 mg) and product from b) (0.5 mg) and c) (0.5 mg) in a vial.
- the mixture was sonicated for 5 min and then heated at 80 ° C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water.
- MALDI mass spectrometry showed no detectable level of compound from b) and c) in the final wash solution.
- a solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5 mg) and product from c) (0.5 mg) and d) (0.5 mg) in a vial.
- the mixture was sonicated for 5 min and then heated at 80° C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water.
- MALDI mass spectrometry showed no detectable level of compound from c and d) in the final wash solution.
- This example is directed at the preparation of functionalised microbubbles with non-specific affinity for a multiplicity of cell surface molecules.
- a solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 4.5 mg) and product from a) (0.5 mg) in a vial.
- the mixture was sonicated for 5 min and then heated at 80 ° C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water.
- MALDI mass spectrometry showed no detectable level of compound from a) in the final wash solution.
- microbubbles were evaluated using the in vitro flow assay described in example 1 c). A gradual accumulation of the microbubbles on the cells took place which was dependant on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- microbubbles were evaluated using the in vitro flow assay described in example 1 c). A gradual accumulation of the microbubbles on the cells took place which was dependant on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- the human endothelial cell line ECV 304 derived from a normal umbilical cord (ATCC CRL-1998) was cultures in Nunc culture flasks (chutney 153732) in RPMI 1640 medium (Bio Whitaker) to which L-Glutamine 200 mM, Penicillin/Streptomycin (10.000 U/ml and 10.00 mcg/ml) and 10% Fetal Calf Serum (Hyclone Lot no AFE 5183) were added. The cells were subcultured following trypsination with a split ratio of 1:5 to 1:7 when reaching confluence. 2 mill.
- This example is directed at the preparation of targeted microbubbles for gene transfer.
- DSPS 4,5 mg
- lipopeptide from 17 b 0.5 mg
- DSPS lipopeptide from 17 b
- 0.8 ml propyleneglycol/glycerol 4%) in water was added.
- the solution was heated at 80° C. for 5 minutes and shaken.
- the solution was then cooled to ambient temperature and the headspace flushed with perfluorobutane.
- the vials were shaken on a Capmix (Espe Capmix, 4450 oscillations/min) for 45 seconds and put on a roller table for 5 minutes.
- the content of the vials were mixed and the sample washed by centrifugation at 2000 rpm for 5 minutes.
- the infranatant was removed and the same volume of distilled water added.
- the washing procedure was repeated once.
- poly-L-lysine HBr (Sigma, 20.6 mg) was dissolved in 2 mL water then an aliquot (0.4 mL) made up to 2 mL water. To 1.2 mL of the diluted poly-L-lysine solution was added 0.12 mL of the DSPS-lipopeptide bubble suspension. Following incubation excess polylysine was removed by extensive washing with water.
- Endothelial cells (ECV 304) were cultured in 6 well plates to a uniform subconfluent layer.
- a transfection mixture consisting of 5 ⁇ g DNA (an Enhanced Green Fluorescent Protein vector from CLONTECH) and 50 ⁇ l of microbubble suspension from a) in RPMI medium at a final volume of 250 ⁇ l was prepared. The mixture was left standing for 15 min at room temperature then 1 mL of complete RPMI medium added. The medium was removed from the cell culture dish, and the DNA-microbubble mixture added to the cells.
- the cells were incubated in a cell culture incubator (37° C.).
- the cells were further incubated in the cell culture incubator (37 ° C.) for approximately 41 ⁇ 2 hours.
- the medium containing DNA-microbubbles was then removed by aspiration, and 2 ml complete RPMI medium was added.
- the cells were incubated for 40-70 hours before examination. Most of the medium was then removed, and the cells were examined by fluorescence microscopy. The results were compared to the results from control experiments were DNA or DNA-polylysine were added to the cells.
Abstract
Targetable diagnostic and/or therapeutically active agents, e.g. ultrasound contrast agents, comprising a suspension in an aqueous carrier liquid of a reporter comprising gas-containing or gas-generating material, said agent being capable of forming at least two types of binding pairs with a target.
Description
- This invention relates to diagnostic and/or therapeutically active agents, more particularly to diagnostic and/or therapeutically active agents incorporating moieties having affinity for sites and/or structures within the body so that diagnostic imaging and/or therapy of particular locations within the body may be enhanced. Of particular interest are diagnostic agents for use in ultrasound imaging, which are hereinafter referred to as targeted ultrasound contrast agents.
- It is well known that ultrasonic imaging comprises a potentially valuable diagnostic tool, for example in studies of the vascular system, particularly in cardiography, and of tissue microvasculature. A variety of contrast agents has been proposed to enhance the acoustic images so obtained, including suspensions of solid particles, emulsified liquid droplets, gas bubbles and encapsulated gases or liquids. It is generally accepted that low density contrast agents which are easily compressible are particularly efficient in terms of the acoustic backscatter they generate, and considerable interest has therefore been shown in the preparation of gas-containing and gas-generating systems.
- Gas-containing contrast media are also known to be effective in magnetic resonance (MR) imaging, e.g. as susceptibility contrast agents which will act to reduce MR signal intensity. Oxygen-containing contrast media also represent potentially useful paramagnetic MR contrast agents.
- Furthermore, in the field of x-ray imaging it has been observed that gases such as carbon dioxide may be used as negative oral contrast agents or intravascular contrast agents.
- The use of radioactive gases, e.g. radioactive isotopes of inert gases such as xenon, has also been proposed in scintigraphy, for example for blood pool imaging.
- Targeted ultrasound contrast agents may be regarded as comprising (i) a reporter moiety capable of interacting with ultrasound irradiation to generate a detectable signal; (ii) one or more vectors having affinity for particular target sites and/or structures within the body, e.g. for specific cells or areas of pathology; and (iii) one or more linkers connecting said reporter and vector(s), in the event that these are not directly joined.
- The molecules and/or structure to which the contrast agent is intended to bind will hereinafter be referred to as the target. In order to obtain specific imaging of a selected region/structure in the body the target must be present and available in this region/structure. Ideally it will be expressed only in the region of interest, but usually will also be present at other locations in the body, creating possible background problems. The target may either be a defined molecular species (i.e. a target molecule) or an unknown molecule or more complex structure (i.e. a target structure) which is present in the area to be imaged, and is able to bind specifically or selectively to a given vector molecule.
- The vector is attached to the reporter moiety in order to bind these moieties to the region/structure to be imaged. The vector may bind specifically to a chosen target, or it may bind only selectively, having affinty also for a limited number of other molecules/structures, again creating possible background problems.
- There is a limited body of prior art relating to targeted ultrasound contrast agents. Thus, for example, U.S. Pat No. 5,531,980 is directed to systems in which the reporter comprises an aqueous suspension of air or gas microbubbles stabilised by one or more film-forming surfactants present at least partially in lamellar or laminar form, said surfactant(s) being bound to one or more vectors comprising “bioactive species designed for specific targeting purposes”. It is stated that the microbubbles are not directly encapsulated by surfactant material but rather that this is incorporated in liquid-filled liposomes which stabilise the microbubbles. It will be appreciated that lamellar or laminar surfactant material such as phospholipids present in such liposomes will inevitably be present in the form of one or more lipid bilayers with the lipophilic tails “back-to-back” and the hydrophilic heads both inside and outside (see e.g. Schneider, M. on “Liposomes as drug carriers: 10 years of research” in Drug targeting, Nyon, Switzerland, 3-5 Oct. 1984, Buri, P. and Gumma, A. (Ed), Elsevier, Amsterdam 1984).
- EP-A-0727225 describes targeted ultrasound contrast agents in which the reporter comprises a chemical having a sufficient vapour pressure such that a proportion of it is a gas at the body temperature of the subject. This chemical is associated with a surfactant or albumin carrier which includes a protein-, peptide- or carbohydrate-based cell adhesion molecule ligand as vector. The receptor moieties in such contrast agents correspond to the phase shift colloid systems described in WO-A-9416739; it is now recognised that administration of such phase shift colloids may lead to generation of microbubbles which grow uncontrollably, possibly to the extent where they cause potentially dangerous embolisation of, for example, the myocardial vasculature and brain (see e.g. Schwarz, Advances in Echo-Contrast [1994(3)], pp 48-49).
- WO-A-9320802 proposes that tissue-specific ultrasonic image enhancement may be achieved using acoustically reflective oligolamellar liposomes conjugated to tissue-specific ligands such as antibodies, peptides, lectins etc. The liposomes are deliberately chosen to be devoid of gas and so will not have the advantageous echogenic properties of gas-based ultrasound contrast agents. Further references to this technology, e.g. in targeting to fibrin, thrombi and atherosclerotic areas are found in publications by Alkanonyuksel, H. et al. in J. Pharm. Sci. (1996) 85(5), 486-490; J. Am. Coll. Cardiol. (1996) 27(2) Suppl A, 298A; and Circulation, 68 Sci. Sessions, Anaheim 13-16 Nov. 1995.
- There is also a number of publications concerning ultrasound contrast agents which refer in passing to possible use of monoclonal antibodies as vectors without giving significant practical detail and/or to reporters comprising materials which may be taken up by the reticuloendothelial system and thereby permit image enhancement of organs such as the liver—see, for example WO-A-9300933, WO-A-9401140, WO-A-9408627, WO-A-9428874, U.S. Pat No. 5,088,499, U.S. Pat No. 5,348,016 and U.S. Pat No. 5,469,854. In general these prior art targeted contrast agents are intended to enhance contrast at specific sites in the body, for example tumour cells, by using one vector to bind strongly to one target, in order to achieve concentration at the target cells. In contrast to this principle of using one vector to bind with high affinity to one target, the present invention is based in part on the finding that diagnostic and/or therapeutically active agents with more favourable properties may be obtained by use of multiple kinds of vector-target interactions (e.g. involving agents associated with a plurality of different vectors and/or with one or more vectors having affinity for different targets on the same or different cell types). In this way, binding of gas-containing and gas-generating diagnostic and/or therapeutic agents may, for example, be obtained by forming multiple binding pairs between one vector with specificity for more than one receptor or between more than one vector with affinity for one or more types of target, with either low or high affinities. Such multiple binding of the vector-conjugated agent to one or more target molecules/structures may result in advantageous targeting properties, for example by enhancing target specificity and/or by distinguishing interactions at a desired target area from background interactions with lower levels of molecules/structures similar to target expressed elsewhere in the body.
- It is well known to use one vector binding with high affinity to one target. The present invention, however, is based on the finding that the desired binding of gas-containing and gas-generating diagnostic and/or therapeutic agents may be obtained by forming multiple binding pairs with low affinity between one type of vector and one type of target, or by forming multiple binding pairs between one or more types of vectors and one or more types of target, with either low or high affinities. Thus multiple binding of the vector conjugated agent to one or more target molecules/structures may have advantageous targeting properties, for example in enhancing target specificity and/or in distinguishing interactions at a desired target area from background interactions with lower levels of molecules/structures similar to target expressed elsewhere in the body.
- Thus according to one aspect of the present invention there is provided a targetable diagnostic and/or therapeutically active agent, e.g. an ultrasound contrast agent, comprising a suspension in an aqueous carrier liquid, e.g. an injectable carrier liquid, of a reporter comprising gas-containing or gas-generating material characterised in that said agent is capable of forming at least two types of binding pairs, e.g. being conjugated to at least two vectors or to one vector capable of binding to at least two binding sites.
- One advantageous embodiment of the invention is based on the additional finding that limited adhesion to targets is a highly useful property of diagnostic and/or therapeutically active agents, which property may be achieved using vectors giving temporary retention rather than fixed adhesion to a target. Thus such agents, rather than being fixedly retained at specific sites, may for example effectively exhibit a form of retarded flow along the vascular endothelium by virtue of their transient interactions with endothelial cells. Such agents may thus become concentrated on the walls of blood vessels, in the case of ultrasound contrast agents providing enhanced echogenicity thereof relative to the bulk of the bloodstream, which is devoid of anatomical features. They therefore may permit enhanced imaging of the capillary system, including the microvasculature, and so may facilitate distinction between normal and inadequately perfused tissue, e.g. in the heart, and may also be useful in visualising structures such as Kupffer cells, thrombi and atherosclerotic lesions or for visualising neo-vascularized and inflamed tissue areas. The present invention is well suited to imaging changes occurring in normal blood vessels which are situated in areas of tissue necrosis.
- It will be appreciated that binding affinities are dependent on numbers of interactions as well as their strength. The density of vector molecules at the surface of the reporter units may therefore be selected so as appropriately to adjust the degree of interactions between particular agents and targets.
- The term multiple-specificity is also used to describe an injectable carrier liquid, of gas-containing or gas-generating material composed of one or more vectors with a specificity for one or more cellular surface receptors while at the same time comprising a second element with specificity for a substrate or receptor system binding to which induces a therapeutic response. Thus included within the scope of the present invention are multiple-specific imaging agents comprising a targeting vector, such as the anti-fibrin antibody described by Lanza et al. (Circulation, (1996) 94 (12),pp 3334), annexin V atherosclerotic plaque binding peptides such as YRALVDTLK, or any other vector known to associate with fibrin clots, in combination with a drug or enzyme with fibrinolytic activity such as streptokinase, plasminogen activator (tPA), urokinase (uPA) or prourokinase (scuPA) resulting in a localised therapeutic antithrombotic effect. This invention is also extended to include vectors with increased specificity for tumour cells in combination with vectors or drug molecules functioning as chemotherapeutic agents capable of inhibiting tumour growth.
- It is well known that many, if not all, target molecules are not expressed exclusively at target sites; a common situation is that such molecules are over-expressed by target cells or at a target structure but are also expressed at lower levels elsewhere in the body. The use of reporters carrying a multiplicity of vectors with relatively low affinity for the target may be advantageous in this situation, since the reporter will then tend to concentrate in regions of high target density which permit multiple (and therefore strong) binding to the reporter (e.g. a gas-containing agent incorporating the vectors folic acid and glutathione for multiple-specific binding to folic acid receptors and glutathione-S-trasferase receptors respectively which are over-expressed as tumour cells). Areas of low target density, on the other hand, will not provide sufficient interaction with such low affinity vectors to bind the target. In such embodiments of the invention, low affinity vectors may be regarded as having an association constant Kafor interaction with a target molecule or structure of less than 108M−1, e.g. less than 107 M−1, preferably less than 106M−1. A further embodiment of this invention is thus based on the finding that the desired binding of gas-containing and gas-generating diagnostic and/or therapeutic agents may be obtained by forming binding pairs with low affinity between more than one type of vector and one or more type of target. Multiple vectors may therfore be used to increase specificity, so that the reporter will bind only to target cells or structures expressing a particular combination of target molecules.
- It may also be useful to select a plurality of vectors which bind to different parts, e.g. epitopes, of a target structure in order to give increased binding strength. This may be particularly advantageous when the target density is low.
- Products comprising two or more vectors with different specificities, i.e. which bind to different target molecules on different cells, may advantageously be used as “general purpose” agents for detection of a range of diseases, e.g. different forms of cancer. Thus, for example, the use of such agents may enable detection of metastases, which are often heterogeneous with respect to expression of target molecules (i.e. antigens).
- Within the context of the present invention, the reporter unit will usually remain attached to the vectors. In another type of targeting procedure, sometimes called pre-targeting, the vector (often, a monoclonal antibody) is administered alone; subsequently, the reporter is administered, coupled to a moiety which is capable of specifically binding the vector molecule (when the vector is an antibody, the reporter may be coupled to an immunoglobulin-binding molecule, such as protein A or an anti-immunoglobulin antibody). An advantage of this protocol is that time may be allowed for elimination of the vector molecules that do not bind their targets, substantially reducing the background problems that are connected with the presence of an excess of reporter-vector conjugate. Within the context of the present invention, pre-targeting with one specific vector might be envisaged, followed by reporter units that are coupled to another vector and a moiety which binds the first vector.
- Within the context of the present invention, in some cases and in particular for the assessment of blood perfusion rates in defined areas, for example in myocardium, it is of interest to measure the rate at which ultrasound contrast agents bound to the target are displaced or released from the target. This can be achieved in a controlled fashion by subsequent administration of a vector or other agent able to displace or release the contrast agent from the target.
- Vectors useful in accordance with the invention include ligands for cell adhesion proteins, as well as cell adhesion proteins themselves where these have corresponding ligands on endothelial cell surfaces. Examples of cell adhesion proteins include integrins, most of which bind the Arg-Gly-Asp (RGD) amino acid sequence. If desired, the vector may be targeted to specific cell adhesion proteins expressed mainly on activated endothelial cells such as are found at or close to sites of inflammation or other pathological responses. Other vectors which may be used include proteins and peptides that bind to cell-surface proteoglycans, which are complexes of proteins and sulphated polysaccarides found on most cells, including endothelial cells. Such proteoglycans contribute to the negative surface charge of all nucleated cells from vertebrate animals; this charge may also be exploited in accordance with the invention by using positively charged vectors, e.g. comprising cationic lipids, which will interact electrostatically with the endothelial surface.
- A further aspect of the present invention is for example where a vector or vectors is attached to the reporter or included non-covalently into the reporter in a manner where the said vector or vectors is not readily exposed to the targets or receptors. Increased tissue specificity may therefore be achieved by applying an additional process to expose the vectors, e.g. the agent is exposed after administration to external ultrasound to change the diffusibility of the moieties containing the vectors.
- The reporter may be in any convenient form, for example being any appropriate gas-containing or gas-generating ultrasound contrast agent formulation. Representative examples of such formulations include microbubbles of gas stabilised (e.g. at least partially encapsulated) by a coalescence-resistant surface membrane (for example gelatin, e.g. as described in WO-A-8002365), a filmogenic protein (for example an albumin such as human serum albumin, e.g. as described in U.S. Pat No. 4,718,433, U.S. Pat No. 4,774,958, U.S. Pat No. 4,844,882, EP-A-0359246, WO-A-9112823, WO-A-9205806, WO-A-9217213, WO-A-9406477 or WO-A-9501187), a polymer material (for example a synthetic biodegradable polymer as described in EP-A-0398935, an elastic interfacial synthetic polymer membrane as described in EP-A-0458745, a microparticulate biodegradable polyaldehyde as described in EP-A-0441468, a microparticulate N-dicarboxylic acid derivative of a polyamino acid—polycyclic imide as described in EP-A-0458079, or a biodegradable polymer as described in WO-A-9317718 or WO-A-9607434), a non-polymeric and non-polymerisable wall-forming material (for example as described in WO-A-9521631), or a surfactant (for example a polyoxyethylene-polyoxypropylene block copolymer surfactant such as a Pluronic, a polymer surfactant as described in WO-A-9506518, or a film-forming surfactant such as a phospholipid, e.g. as described in WO-A-9211873, WO-A-9217212, WO-A-9222247, WO-A-9428780 or WO-A-9503835).
- Other useful gas-containing contrast agent formulations include gas-containing solid systems, for example microparticles (especially aggregates of microparticles) having gas contained therewithin or otherwise associated therewith (for example being adsorbed on the surface thereof and/or contained within voids, cavities or pores therein, e.g. as described in EP-A-0122624, EP-A-0123235, EP-A-0365467, WO-A-9221382, WO-A-9300930, WO-A-9313802, WO-A-9313808 or WO-A-9313809). It will be appreciated that the echogenicity of such microparticulate contrast agents may derive directly from the contained/associated gas and/or from gas (e.g. microbubbles) liberated from the solid material (e.g. upon dissolution of the microparticulate structure).
- The disclosures of all of the above-described documents relating to gas-containing contrast agent formulations are incorporated herein by reference.
- Gas microbubbles and other gas-containing materials such as microparticles preferably have an initial average size not exceeding 10 μm (e.g. of 7 μm or less) in order to permit their free passage through the pulmonary system following administration, e.g. by intravenous injection.
- Where phospholipid-containing compositions are employed in accordance with the invention, e.g. in the form of phospholipid-stabilised gas microbubbles, representative examples of useful phospholipids include lecithins (i.e. phosphatidylcholines), for example natural lecithins such as egg yolk lecithin or soya bean lecithin and synthetic or semisynthetic lecithins such as dimyristoylphosphatidylcholine, dipalmitoylphosphatidylcholine or distearoylphosphatidylcholine; phosphatidic acids; phosphatidylethanolamines; phosphatidylserines; phosphatidylglycerols; phosphatidylinositols; cardiolipins; sphingomyelins; fluorinated analogues of any of the foregoing; mixtures of any of the foregoing and mixtures with other lipids such as cholesterol. The use of phospholipids predominantly (e.g. at least 75%) comprising molecules individually bearing net overall charge, e.g. negative charge, for example as in naturally occurring (e.g. soya bean or egg yolk derived), semisynthetic (e.g. partially or fully hydrogenated) and synthetic phosphatidylserines, phosphatidylglycerols, phosphatidylinositols, phosphatidic acids and/or cardiolipins, may be particularly advantageous.
- Other exemplary lipids which may be used to prepare gas-containing contrast agents include fatty acids, stearic acid, palmitic acid, 2-n-hexadecylstearic acid, oleic acid and other acid containing lipid structures. These lipid structures are considered particularly interesting when coupled by amide bond formation to amino acids containing one or more amino groups. The resulting lipid modified amino acids (e.g. dipalmitoyllysine, distearoyl-2,3-diaminopropionic acid) are considered useful precursors for the attachment of functionalised spacer elements featuring coupling sites for conjugation of one or more vector molecules.
- A further extension of this invention relates to the synthesis of lipopeptide structures comprising a lipid reporter attached to a linker portion (e.g. PEG, polyamino acid, alkylhalide etc) the said linker being suitably functionalised for coupling to one or more vector molecules. A particular preference is the inclusion of a positively charged linker element (e.g. two or more lysine residues) for anchoring of the reporter element in the microbubble through electrostatic interaction with the negatively charged membrane. Multiple-specific targeting is achievable by mixing and ‘doping’ of phospholipid gas-containing structures with one or more targeted lipopeptide sequences. Multiple-specificity can also be achieved by assembling more than one vector on a branched lysine core structure such as those described by Tam et. al. (Proc. Natl. Acad. Sci. USA, 1989, 86, 9084) or by incorporating multiple vectors in a linear sequence. Multiple-specificity can also be achieved using lipopeptides or phospholipids comprising combinatorial libraries synthesised by chemical synthesis as described by Lowe (Combinatorial Chemistry, Chemical Society Reviews, 1995, 309-317).
- Also within the scope of this invention are functionalised microbubbles carrying one or more reactive groups for non-specific reaction with receptor molecules located on cell surfaces. Microbubbles comprising a thiol moiety,for example, can bind to cell surface receptors via disulphide exchange reactions. The reversible nature of this covalent bond means that bubble flow can be controlled by altering the redox environment. Similarly ‘activated’ microbubbles of membranes comprising active esters such as N-hydroxysuccinimide esters can be used to modify amino groups found on a multiplicity of cell surface molecules.
- Representative examples of gas-containing microparticulate materials which may be useful in accordance with the invention include carbohydrates (for example hexoses such as glucose, fructose or galactose; disaccharides such as sucrose, lactose or maltose; pentoses such as arabinose, xylose or ribose; α-, β- and γ-cyclodextrins; polysaccharides such as starch, hydroxyethyl starch, amylose, amylopectin, glycogen, inulin, pulullan, dextran, carboxymethyl dextran, deXtran phosphate, ketodextran, aminoethyldextran, alginates, chitin, chitosan, hyaluronic acid or heparin; and sugar alcohols, including alditols such as mannitol or sorbitol), inorganic salts (e.g. sodium chloride), organic salts (e.g. sodium citrate, sodium acetate or sodium tartrate), X-ray contrast agents (e.g. any of the commercially available carboxylic acid and non-ionic amide contrast agents typically containing at least one 2,4,6-triiodophenyl group having substituents such as carboxyl, carbamoyl, N-alkylcarbamoyl, N-hydroxyalkylcarbamoyl, acylamino, N-alkylacylamino or acylaminomethyl at the 3- and/or 5-positions, as in metrizoic acid, diatrizoic acid, iothalamic acid, ioxaglic acid, iohexol, iopentol, iopamidol, iodixanol, iopromide, metrizamide, iodipamide, meglumine iodipamide, meglumine acetrizoate and meglumine diatrizoate), and polypeptides and proteins (e.g. gelatin or albumin such as human serum albumin).
- Any biocompatible gas may be present in the reporter of contrast agents according to the invention, the term “gas” as used herein including any substances (including mixtures) substantially or completely in gaseous (including vapour) form at the normal human body temperature of 37° C. The gas may thus, for example, comprise air; nitrogen; oxygen; carbon dioxide; hydrogen; an inert gas such as helium, argon, xenon or krypton; a sulphur fluoride such as sulphur hexafluoride, disulphur decafluoride or trifluoromethylsulphur pentafluoride; selenium hexafluoride; an optionally halogenated silane such as methylsilane or dimethylsilane; a low molecular weight hydrocarbon (e.g. containing up to 7 carbon atoms), for example an alkane such as methane, ethane, a propane, a butane or a pentane, a cycloalkane such as cyclopropane, cyclobutane or cyclopentane, an alkene such as ethylene, propene, propadiene or a butene, or an alkyne such as acetylene or propyne; an ether such as dimethyl ether; a ketone; an ester; a halogenated low molecular weight hydrocarbon (e.g. containing up to 7 carbon atoms); or a mixture of any of the foregoing. Advantageously at least some of the halogen atoms in halogenated gases are fluorine atoms; thus biocompatible halogenated hydrocarbon gases may, for example, be selected from bromochlorodifluoromethane, chlorodifluoromethane, dichlorodifluoromethane, bromotrifluoromethane, chlorotrifluoromethane, chloropentafluoroethane, dichlorotetrafluoroethane, chlorotrifluoroethylene, fluoroethylene, ethylfluoride, 1,1-difluoroethane and perfluorocarbons, e.g. perfluoroalkanes such as perfluoromethane, perfluoroethane, perfluoropropanes, perfluorobutanes (e.g. perfluoro-n-butane, optionally in admixture with other isomers such as perfluoro-iso-butane), perfluoropentanes, perfluorohexanes and perfluoroheptanes; perfluoroalkenes such as perfluoropropene, perfluorobutenes-(e.g. perfluorobut-2-ene) and perfluorobutadiene; perfluoroalkynes such as perfluorobut-2-yne; and perfluorocycloalkanes such as perfluorocyclobutane, perfluoromethylcyclobutane, perfluorodimethylcyclobutanes, perfluorotrimethyl-cyclobutanes, perfluorocyclopentane, perfluoromethyl-cyclopentane, perfluorodimethylcyclopentanes, perfluorocyclohexane, perfluoromethylcyclohexane and perfluorocycloheptane. Other halogenated gases include methyl chloride, fluorinated (e.g. perfluorinated) ketones such as perfluoroacetone and fluorinated (e.g. perfluorinated) ethers such as perfluorodiethyl ether. The use of perfluorinated gases, for example sulphur hexafluoride and perfluorocarbons such as perfluoropropane, perfluorobutanes and perfluoropentanes, may be particularly advantageous in view of the recognised high stability in the bloodstream of microbubbles containing such gases.
- The reporter may be made by any convenient process, for example by making gas-containing or gas-generating formulations. Representative examples include the preparation of a suspension of gas microbubbles by contacting a surfactant with gas and mixing them in the presence of an aqueous carrier, as described in WO 9115244; or by atomising a solution or dispersion of a wall-forming material in the presence of a gas in order to obtain hollow microcapsules, as described in EP 512693A1; preparation of solid microspheres by a double emulsion process, as described in U.S. Pat. No. 5,648,095; or a process for forming hollow microcapsules by spray-drying as described in EP 681843A2; or preparing gas-filled liposomes by shaking an aqueous solution comprising a lipid in the presence of a gas as described in U.S. Pat. No. 5,469,854.
- A suitable process for attachment of the desired vector to the reporter comprises a surface modification of the preformed reporter with a suitable linker employing reactive groups on the surface of both the reporter and vector. It may be particularly advantageous physically to mix the reporter material with the vector-containing substance at any step of the process. Such a process will result in incorporation or an attachment of the vector to the reporter. An optional process step may remove the excess of vector not bound to the reporter by washing the gas-containing particles following separation, by for example, floatation. A preferred aspect is the use of lipopeptide structures incorporating functional groups such as thiol, maleimide biotin etc. which can be premixed if desired with other reporter molecules before formation of gas-containing agents. The attachment of vector molecules may be carried out using the linker reagents listed below.
- Linking of a reporter unit to the desired vectors may be achieved by covalent or non-covalent means, usually involving interaction with one or more functional groups located on the reporter and/or vectors. Examples of chemically reactive functional groups which may be employed for this purpose include amino, hydroxyl, sulfhydryl, carboxyl, and carbonyl groups, as well as carbohydrate groups, vicinal diols, thioethers, 2-aminoalcohols, 2-aminothiols, guanidinyl, imidazolyl and phenolic groups.
- Covalent coupling of reporter and vectors may therefore be effected using linking agents containing reactive moieties capable of reaction with such functional groups. Examples of reactive moieties capable of reaction with sulfhydryl groups include α-haloacetyl compounds of the type X—CH2CO— (where X=Br, Cl or I), which show particular reactivity for sulfhydryl groups but which can also be used to modify imidazolyl, thioether, phenol and amino groups as described by Gurd, F. R. N. in Methods Enzymol. (1967) 11, 532. N-Maleimide derivatives are also considered selective towards sulfhydryl groups, but may additionaly be useful in coupling to amino groups under certain conditions. N-maleimides may be incorporated into linking systems for reporter-vector conjugation as described by Kitagawa, T. et al. in Chem. Pharm. Bull. (1981) 29, 1130 or used as polymer crosslinkers for bubble stabilisation as described by Kovacic, P. et al. in J. Am. Chem. Soc. (1959) 81, 1887. Reagents such as 2-iminothiolane, e.g. as described by Traut, R. et al. in Biochemistry (1973) 12, 3266, which introduce a thiol group through conversion of an amino group, may be considered as sulfhydryl reagents if linking occurs through the formation of disulphide bridges. Thus reagents which introduce reactive disulphide bonds into either the reporter or the vectors may be useful, since linking may be brought about by disulphide exchange between the vector and reporter; examples of such reagents include Ellman's reagent (DTNB), 4,4′-dithiodipyridine, methyl-3-nitro-2-pyridyl disulphide and methyl-2-pyridyl disulphide (described by Kimura, T. et al. in Analyt. Biochem. (1982) 122, 271).
- Examples of reactive moieties capable of reaction with amino groups include alkylating and acylating agents. Representative alkylating agents include:
- i) α-haloacetyl compounds, which show specificity towards amino groups in the absence of reactive thiol groups and are of the type X—CH2CO— (where X=Cl, Br or I), e.g. as described by Wong, Y-H. H. in Biochemistry (1979) 24, 5337;
- ii) N-maleimide derivatives, which may react with amino groups either through a Michael type reaction or through acylation by addition to the ring carbonyl group as described by Smyth, D. G. et al. in J. Am. Chem. Soc. (1960) 82, 4600 and Biochem. J. (1964) 91, 589;
- iii) aryl halides such as reactive nitrohaloaromatic compounds;
- iv) alkyl halides as described by McKenzie, J. A. et al. in J. Protein Chem. (1988) 7, 581;
- v) aldehydes and ketones capable of Schiff's base formation with amino groups, the adducts formed usually being stabilised through reduction to give a stable amine;
- vi) epoxide derivatives such as epichlorohydrin and bisoxiranes,which may react with amino, sulfhydryl or phenolic hydroxyl groups;
- vii) chlorine-containing derivatives of s-triazines, which are very reactive towards nucleophiles such as amino, sufhydryl and hydroxy groups;
- viii) aziridines based on s-triazine compounds detailed above, e.g. as described by Ross, W. C. J. in Adv. Cancer Res. (1954) 2, 1, which react with nucleophiles such as amino groups by ring opening;
- ix) squaric acid diethyl esters as described by Tietze, L. F. in Chem. Ber. (1991) 124, 1215; and
- x) α-haloalkyl ethers, which are more reactive alkylating agents than normal alkyl halides because of the activation caused by the ether oxygen atom, e.g. as described by Benneche, T. et al. in Eur. J. Med. Chem. (1993) 28, 463.
- Representative amino-reactive acylating agents include:
- i) isocyanates and isothiocyanates, particularly aromatic derivatives, Which form stable urea and thiourea derivatives respectively and have been used for protein crosslinking as described by Schick, A. F. et al. in J. Biol. Chem. (1961) 236, 2477;
- ii) sulfonyl chlorides, which have been described by Herzig, D. J. et al. in Biopolymers (1964) 2, 349 and which may be useful for the introduction of a fluorescent reporter group into the linker;
- iii) Acid halides;
- iv) Active esters such as nitrophenylesters or N-hydroxysuccinimidyl esters;
- v) acid anhydrides such as mixed, symmetrical or N-carboxyanhydrides;
- vi) other useful reagents for amide bond formation as described by Bodansky, M. et al. in ‘Principles of Peptide Synthesis’ (1984) Springer-Verlag;
- vii) acylazides, e.g. wherein the azide group is generated from a preformed hydrazide derivative using sodium nitrite, e.g. as described by Wetz, K. et al. in Anal. Biochem. (1974) 58, 347;
- viii) azlactones attached to polymers such as bis-acrylamide, e.g. as described by Rasmussen, J. K. in Reactive Polymers (1991) 16, 199; and
- ix) Imidoesters, which form stable amidines on reaction with amino groups, e.g. as described by Hunter, M. J. and Ludwig, M. L. in J. Am. Chem. Soc. (1962) 84, 3491.
- Carbonyl groups such as aldehyde functions may be reacted with weak protein bases at a pH such that nucleophilic protein side-chain functions are protonated. Weak bases include 1,2-aminothiols such as those found in N-terminal cysteine residues, which selectively form stable 5-membered thiazolidine rings with aldehyde groups, e.g. as described by Ratner, S. et al. in J. Am. Chem. Soc. (1937) 59, 200. Other weak bases such as phenyl hydrazones may be used, e.g. as described by Heitzman, H. et al. in Proc. Natl. Acad. Sci. USA (1974) 71, 3537.
- Aldehydes and ketones may also be reacted with amines to form Schiff's bases, which may advantageously be stabilised through reductive amination. Alkoxylamino moieties readily react with ketones and aldehydes to produce stable alkoxamines, e.g. as described by Webb, R. et al. in Bioconjugate Chem. (1990) 1, 96.
- Examples of reactive moieties capable of reaction with carboxyl groups include diazo compounds such as diazoacetate esters and diazoacetamides, which react with high specificity to generate ester groups, e.g. as described by Herriot R. M. in Adv. Protein Chem. (1947) 3, 169. Carboxylic acid modifying reagents such as carbodiimides, which react through O-acylurea formation followed by amide bond formation, may also usefully be employed; linking may be facilitated through addition of an amine or may result in direct vector-receptor coupling. Useful water soluble carbodiimides include 1-cyclohexyl-3-(2-morpholinyl-4-ethyl)carbodiimide (CMC) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), e.g. as described by Zot, H. G. and Puett, D. in J. Biol. Chem. (1989) 264, 15552. Other useful carboxylic acid modifying reagents include isoxazolium derivatives such as Wo6dwards reagent K; chloroformates such as p-nitrophenylchloroformate; carbonyldiimidazoles such as 1,1′-carbonyldiimidazole; and N-carbalkoxydihydroquinolines such as N-(ethoxycarbonyl)-2-ethoxy-1,2-dihydroquinoline.
- Other potentially useful reactive moieties include vicinal diones such as p-phenylenediglyoxal, which may be used to react with guanidinyl groups, e.g. as described by Wagner et al. in Nucleic acid Res. (1978) 5, 4065; and diazonium salts, which may undergo electrophilic substitution reactions, e.g. as described by Ishizaka, K. and Ishizaka T. in J. Immunol. (1960) 85, 163. Bis-diazonium compounds are readily prepared by treatment of aryl diamines with sodium nitrite in acidic solutions. It will be appreciated that functional groups in the reporter and/or vector may if desired be converted to other functional groups prior to reaction, e.g. to confer additional reactivity or selectivity. Examples of methods useful for this purpose include conversion of amines to carboxylic acids using reagents such as dicarboxylic anhydrides; conversion of amines to thiols using reagents such as N-acetylhomocysteine thiolactone, S-acetylmercaptosuccinic anhydride, 2-iminothiolane or thiol-containing succinimidyl derivatives; conversion of thiols to carboxylic acids using reagents such as a-haloacetates; conversion of thiols to amines using reagents such as ethylenimine or 2-bromoethylamine; conversion of carboxylic acids to amines using reagents such as carbodiimides followed by diamines; and conversion of alcohols to thiols using reagents such as tosyl chloride followed by transesterification with thioacetate and hydrolysis to the thiol with sodium acetate.
- Vector-receptor coupling may also be effected using enzymes as zero-length crosslinking agents; thus, for example, transglutaminase, peroxidase and xanthine oxidase have been used to produce crosslinked products. Reverse proteolysis may also be used for crosslinking through amide bond formation.
- Non-covalent vector-receptor coupling may, for example, be effected by electrostatic charge interactions e.g. between a polylysinyl-functionalised reporter and a polyglutamyl-functionalised vector, through chelation in the form of stable metal complexes or through high affinity binding interaction such as avidin/biotin binding. Polylysine, coated non-covalently to the negatively charged membrane surface can also increase non-specifically the affinity of a microbubble for a cell through charge interactions.
- Alternatively, vectors may be coupled to a protein or peptide sequence known to bind phospholipids. In many instances, a single molecule of phospholipid may attach to a protein such as a translocase, while other proteins may attach to surfaces consisting mainly of phospholipid head groups and so may be used to attach vectors to phospholipid microspheres; one example of such a protein is β2-glycoprotein I (Chonn, A., Semple, S. C. and Cullis, P. R., Journal of Biological Chemistry (1995) 270, 25845-25849). Phosphatidylserine-binding proteins have been described, e.g. by Igarashi, K. et al. in Journal of Biological Chemistry 270(49), 29075-29078. Annexins are a class of phospholipid-binding proteins, many of which bind particularly avidly to phosphatidyl-serine (reviewed in Raynal, P. and H. B. Pollard. Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins”. Biochim. Biophys. Acta 1197: 63-93). A conjugate of a vector with such a phosphatidylserine-binding protein may therefore be used to attach the vector to phosphatidylserine-encapsulated microbubbles. When the amino acid sequence of a binding protein is known, the phospholipid-binding portion may be synthesised or isolated and used for conjugation with a vector, thus avoiding the biological activity which may be located elsewhere in the molecule.
- It is also possible to obtain molecules that bind specifically to the surface (or in the “membrane”) of microspheres by direct screening of molecular libraries for microsphere-binding molecules. For example, phage libraries displaying small peptides could be used for such selection. The selection may be made by simply mixing the microspheres and the phage display library and eluting the phages binding to the floating microspheres. If desired, the selection can be done under “physiological conditions” (e.g. in blood) to eliminate peptides which cross-react with blood components. An advantage of this type of selection procedure is that only binding molecules that do not destabilize the microspheres should be selected, since only binding molecules attached to intact floating microspheres will rise to the top. It may also be possible to introduce some kind of “stress” during the selection procedure (e.g. pressure) to ensure that destabilizing binding moieties are not selected. Furthermore the selection could be done under shear conditions e.g. by first letting the phages react with the microspheres and then letting the microspheres pass through a surface coated with anti-phage antibodies under flow conditions. In this way it may be possible to select binders which may resist shear conditions present in vivo. Binding moieties identified in this way may be coupled (chemically via peptide synthesis, or at the DNA-level using recombinant vectors) to a vector molecule, constituting a general tool for attaching any vector molecule to the microspheres.
- A vector which comprises or is coupled to a peptide or lipopeptide linker which contains a element capable of mediating membrane insertion may also be useful. One example is described by Leenhouts, J. M. et al. in Febs Letters (1995) 370(3), 189-192. Non-bioactive molecules consisting of known membrane insertion anchor/signal groups may also be used as vectors for certain applications, an example being the H1 hydrophobic segment from the Na, K-ATPase α-subunit described by Xie, Y. and Morimoto, T. in J. Biol. Chem. (1995) 270(20), 11985-11991. The anchor group may also be fatty acid(s) or cholesterol.
- Coupling may also be effected using avidin or streptavidin, which have four high affinity binding sites for biotin. Avidin may therefore be used to conjugate vector to reporter if both vector and reporter are biotinylated. Examples are described by Bayer, E. A. and Wilchek, M. in Methods Biochem. Anal. (1980) 26, 1. This method may also be extended to include linking of reporter to reporter, a process which may encourage bubble association and consequent potentially increased echogenicity.
- Non-covalent coupling may also utilise the bifunctional nature of bispecific immunoglobulins. These molecules can specifically bind two antigens, thus linking them. For example, either bispecific IgG or chemically engineered bispecific F(ab)′2 fragments may be used as linking agents. Heterobifunctional bispecific antibodies have also been reported for linking two different antigens, e.g. as described by Bode, C. et al. in J. Biol. Chem. (1989) 264, 944 and by Staerz, U. D. et al. in Proc. Natl. Acad. Sci. USA (1986) 83, 1453. Similarly, any reporter and/or vector containing two or more antigenic determinants (e.g. as described by Chen, Aa et al. in Am. J. Pathol. (1988) 130, 216) may crosslink antibody molecules and lead to formation of multi-bubble cross-linked assemblies of potentially increased echogenicity.
- So-called zero-length linking agents, which induce direct covalent joining of two reactive chemical groups without introducing additional linking material (e.g. as in amide bond formation induced using carbodiimides or enzymatically) may, if desired, be used, as may agents such as biotin/avidin systems which induce non-covalent reporter-vector linking and agents which induce hydrophobic or electrostatic interactions.
- Most commonly, however, the linking agent will comprise two or more reactive moieties, e.g. as described above, connected by a spacer element. The presence of such a spacer permits bifunctional linkers to react with specific functional groups within a molecule or between two different molecules, resulting in a bond between these two components and introducing extrinsic linker-derived material into the reporter-vector conjugate. The reactive moieties in a linking agent may be the same (homobifunctional agents) or different (heterobifunctional agents or, where several dissimilar reactive moieties are present, heteromultifunctional agents), providing a diversity of potential reagents that may bring about covalent bonding between any chemical species, either intramolecularly or intermolecularly.
- The nature of extrinsic material introduced by the linking agent may have a critical bearing on the targeting ability and general stability of the ultimate product. Thus it may be desirable to introduce labile linkages, e.g. containing spacer arms which are biodegradable or chemically sensitive or which incorporate enzymatic cleavage sites. Alternatively the spacer may include polymeric components, e.g. to act as surfactants and enhance bubble stability. The spacer may also contain reactive moieties, e.g. as described above to enhance surface crosslinking, or it may contain a tracer element such as a fluorescent probe, spin label or radioactive material.
- Spacer elements may typically consist of aliphatic chains which effectively separate the reactive moieties of the linker by distances of between 5 and 30 Å. They may also comprise macromolecular structures such as poly(ethylene glycols). Such polymeric structures, hereinafter referred to as PEGS, are simple, neutral polyethers which have been given much attention in biotechnical and biomedical applications (see e.g. Milton Harris, J. (ed) “Poly(ethylene glycol) chemistry, biotechnical and biomedical applications” Plenum Press, New York, 1992). PEGs are soluble in most solvents, including water, and are highly hydrated in aqueous environments, with two or three water molecules bound to each ethylene glycol segment; this has the effect of preventing adsorption either of other polymers or of proteins onto PEG-modified surfaces. PEGs are known to be nontoxic and not to harm active proteins or cells, whilst covalently linked PEGs are known to be non-immunogenic and non-antigenic. Furthermore, PEGs may readily be modified and bound to other molecules with only little effect on their chemistry. Their advantageous solubility and biological properties are apparent from the many possible uses of PEGs and copolymers thereof, including block copolymers such as PEG-polyurethanes and PEG-polypropylenes.
- Appropriate molecular weights for PEG spacers used in accordance with the invention may, for example, be between 120 Daltons and 20 kDaltons.
- The major mechanism for uptake of particles by the cells of the reticuloendothelial system (RES) is opsonisation by plasma proteins in blood; these mark foreign particles which are then taken up by the RES. The biological properties of PEG spacer elements used in accordance with the invention may serve to increase contrast agent circulation time in a similar manner to that observed for PEGylated liposomes (see e.g. Klibanov, A. L. et al. in FEBS Letters (1990) 268, 235-237 and Blume, G. and Cevc, G. in Biochim. Biophys. Acta (1990) 1029, 91-97).
- Other potentially useful protein modifications which can be made to vectors include partial or complete deglycosidation by neuraminidase, endoglycosydases or periodate, since deglycosidation often results in less uptake by liver, spleen, macrophages etc., whereas neo-glycosylation of proteins often results in increased uptake by the liver and macrophages); preparation of truncated forms by proteolytic cleavage, leading to reduced size and shorter half life in circulation; and cationisation, e.g. as described by Kumagi et al. in J. Biol. Chem. (1987) 262, 15214-15219; Triguero et al. in Proc. Natl. Acad. Sci. USA (1989) 86, 4761-4765; Pardridge et al. in J. Pharmacol. Exp. Therap. (1989) 251, 821-826 and Pardridge and Boado, Febs Lett. (1991) 288, 30-32.
- Increased coupling efficiency to areas of interest may also be achieved using antibodies bound to the terminii of PEG spacers (see e.g. Maruyama, K. et al. in Biochim. Biophys. Acta (1995) 1234, 74-80 and Hansen, C. B. et al. in Biochim. Biophys. Acta (1995) 1239, 133-144).
- In some instances it is considered advantageous to include a PEG component as a stabiliser in conjunction with a vector or vectors or directly to the reporter in the same molecule where the PEG does not serve as a spacer.
- Other representative spacer elements include structural-type polysaccharides such as polygalacturonic acid, glycosaminoglycans, heparinoids, cellulose and marine polysaccharides such as alginates, chitosans and carrageenans; storage-type polysaccharides such as starch, glycogen, dextran and aminodextrans; polyamino acids and methyl and ethyl esters thereof, as in homo- and co-polymers of lysine, glutamic acid and aspartic acid; and polypeptides, oligonucleotides and oligosaccharides, which may or may not contain enzyme cleavage sites.
- In general, spacer elements may contain cleavable groups such as vicinal glycol, azo, sulfone, ester, thioester or disulphide groups. Spacers containing biodegradable methylene diester or diamide groups of formula
—(Z)m.Y.X.C(R1R2).X.Y.(Z)n—
[where X and Z are selected from —O—, —S—, and —NR— (where R is hydrogen or an organic group); each Y is a carbonyl, thiocarbonyl, sulphonyl, phosphoryl or similar acid-forming group: m and n are each zero or 1; and R1 and R2are each hydrogen, an organic group or a group —X.Y.(Z)m—, or together form a divalent organic group] may also be useful; as discussed in, for example, WO-A-9217436 such groups are readily biodegraded in the presence of esterases, e.g. in vivo, but are stable in the absence of such enzymes. They may therefore advantageously be linked to therapeutic agents to permit slow release thereof. - Poly[N-(2-hydroxyethyl)methacrylamides] are potentially useful spacer materials by virtue of their low degree of interaction with cells and tissues (see e.g. Volfová, I., Ríhová, B. and V. R. and Vetvicka, P. in J. Bioact. Comp. Polymers (1992) 7, 175-190). Work on a similar polymer consisting mainly of the closely related 2-hydroxypropyl derivative showed that it was endocytosed by the mononuclear phagocyte system only to a rather low extent (see Goddard, P., Williamson, I., Bron, J., Hutchkinson, L. E., Nicholls, J. and Petrak, K. in J. Bioct. Compat. Polym. (1991) 6, 4-24. ).
- Other potentially useful poymeric spacer materials include:
- i) copolymers of methyl methacrylate with methacrylic acid; these may be erodible (see Lee, P. I. in Pharm. Res. (1993) 10, 980) and the carboxylate substituents may cause a higher degree of swelling than with neutral polymers;
- ii) block copolymers of polymethacrylates with biodegradable polyesters (see e.g. San Roman, J. and Guillen-Garcia, P. in Biomaterials (1991) 12, 236-241);
- iii) cyanoacrylates, i.e. polymers of esters of 2-cyanoacrylic acid—these are biodegradable and have been used in the form of nanoparticles for selective drug delivery (see Forestier, F., Gerrier, P., Chaumard, C., Quero, A. M., Couvreur, P. and Labarre, C. in J. Antimicrob. Chemoter. (1992) 30, 173-179);
- iv) polyvinyl alcohols, which are water-soluble and generally regarded as biocompatible (see e.g. Langer, R. in J. Control. Release (1991) 16, 53-60);
- v) copolymers of vinyl methyl ether with maleic anhydride, which have been stated to be bioerodible (see Finne, U., Hannus, M. and Urtti, A. in Int. J. Pharm. (1992) 78. 237-241);
- vi) polyvinylpyrrolidones, e.g. with molecular weight less than about 25,000, which are rapidly filtered by the kidneys (see Hespe, W., Meier, A. M. and Blankwater, Y. M. in Arzeim.-Forsch./Drug Res. (1977) 27, 1158-1162);
- vii) polymers and copolymers of short-chain aliphatic hydroxyacids such as glycolic, lactic, butyric, valeric and caproic acids (see e.g. Carli, F. in Chim. Ind. (Milan) (1993) 75, 494-9), including copolymers which incorporate aromatic hydroxyacids in order to increase their degradation rate (see Imasaki, K., Yoshida, M., Fukuzaki, H., Asano, M., Kumakura, M., Mashimo, T., Yamanaka, H. and Nagai. T. in Int. J. Pharm. (1992) 81, 31-38);
- viii) polyesters consisting of alternating units of ethylene glycol and terephthalic acid, e.g. DacronR, which are non-degradable but highly biocompatible;
- ix) block copolymers comprising biodegradable segments of aliphatic hydroxyacid polymers (see e.g. Younes, H., Nataf, P. R., Cohn, D., Appelbaum, Y. J., Pizov, G. and Uretzky, G. in Biomater. Artif. Cells Artif. Organs (1988) 16, 705-719), for instance in conjunction with polyurethanes (see Kobayashi, H., Hyon, S. H. and Ikada, Y. in “Water-curable and biodegradable prepolymers”—J. Biomed. Mater. Res. (1991) 25, 1481-1494);
- x) polyurethanes, which are known to be well-tolerated in implants, and which may be combined with flexible “soft” segments, e.g. comprising poly(tetra methylene glycol), poly(propylene glycol) or poly(ethylene glycol)) and aromatic “hard” segments, e.g. comprising 4,4′-methylenebis(phenylene isocyanate) (see e.g. Ratner, B. D., Johnston, A. B. and Lenk, T. J. in J. Biomed. Mater. Res: Applied Biomaterials (1987) 21, 59-90; Sa Da Costa, V. et al. in J. Coll. Interface Sci. (1981) 80, 445-452 and Affrossman, S. et al. in Clinical Materials (1991) 8, 25-31);
- xi) poly(1,4-dioxan-2-ones), which may be regarded as biodegradable esters in view of their hydrolysable ester linkages (see e.g. Song, C. X., Cui, X. M. and Schindler, A. in Med. Biol. Eng. Comput. (1993) 31, S147-150), and which may include glycolide units to improve their absorbability (see Bezwada, R. S., Shalaby, S. W. and Newman, H. D. J. in Agricultural and synthetic polymers: Biodegradability and utilization (1990) (ed Glass, J. E. and Swift, G.), 167-174—ACS symposium Series, #433, Washington D.C., U.S.A.—American Chemical Society);
- xii) polyanhydrides such as copolymers of sebacic acid (octanedioic acid) with bis(4-carboxy-phenoxy)propane, which have been shown in rabbit studies (see Brem, H., Kader, A., Epstein, J. I., Tamargo, R. J., Domb, A., Langer, R. and Leong, K. W. in Sel. Cancer Ther. (1989) 5, 55-65) and rat studies (see Tamargo, R. J., Epstein, J. I., Reinhard, C. S., Chasin, M. and Brem, H. in J. Biomed. Mater. Res. (1989) 23, 253-266) to be useful for controlled release of drugs in the brain without evident toxic effects;
- xiii) biodegradable polymers containing ortho-ester groups, which have been employed for controlled release in vivo (see Maa, Y. F. and Heller, J. in J. Control. Release (1990) 14, 21-28); and
- xiv) polyphosphazenes, which are inorganic polymers consisting of alternate phosphorus and nitrogen atoms (see Crommen, J. H., Vandorpe, J. and Schacht, E. H. in J. Control. Release (1993) 24, 167-180).
- The following tables list linking agents and agents for protein modification which may be useful in preparing targetable contrast agents in accordance with the invention.
- Heterobifunctional Linking Agents
Linking agent Reactivity 1 Reactivity 2 Comments ABH carbohydrate photoreactive ANB-NOS —NH2 photoreactive APDP(1) —SH photoreactive iodinable disulphide linker APG —NH2 photoreactive reacts selectively with Arg at pH 7-8 ASIB(1) —SH photoreactive iodinable ASBA(1) —COOH photoreactive iodinable EDC —NH2 —COOH zero-length linker GMBS —NH2 —SH sulfo-GMBS —NH2 —SH water-soluble HSAB —NH2 photoreactive sulfo-HSAB —NH2 photoreactive water-soluble MBS —NH2 —SH sulfo-MBS —NH2 —SH water-soluble M2C2H carbohydrate —SH MPBH carbohydrate —SH NHS-ASA(1) —NH2 photoreactive iodinable sulfo-NHS- —NH2 photoreactive water-soluble, ASA(1) iodinable sulfo-NHS-LC- —NH2 photoreactive water-soluble, ASA(1) iodinable PDPH carbohydrate —SH disulphide linker PNP-DTP —NH2 photoreactive SADP —NH2 photoreactive disulphide linker sulfo-SADP —NH2 photoreactive water-soluble disulphide linker SAED —NH2 photoreactive disulphide linker SAND —NH2 photoreactive water-soluble disulphide linker SANPAH —NH2 photoreactive sulfo-SANPAH —NH2 photoreactive water-soluble SASD(1) —NH2 photoreactive water-soluble iodinable disulphide linker SIAB —NH2 —SH sulfo-SIAB —NH2 —SH water-soluble SMCC —NH2 —SH sulfo-SMCC —NH2 —SH water-soluble SMPB —NH2 —SH sulfo-SMPB —NH2 —SH water-soluble SMPT —NH2 —SH sulfo-LC-SMPT —NH2 —SH water-soluble SPDP —NH2 —SH sulfo-SPDP —NH2 —SH water-soluble sulfo-LC-SPDP —NH2 —SH water-soluble sulfo-SAMCA(2) —NH2 photoreactive sulfo-SAPB —NH2 photoreactive water-soluble
Notes:
(1) = iodinable;
(2) = fluorescent
- Homobifunctional Linking Agents
Linking agent Reactivity Comments 0 —NH2 BMH —SH BASED (1) photoreactive iodinable disulphide linker BSCOES —NH2 sulfo-BSCOES —NH2 water-soluble DFDNB —NH2 DMA —NH2 DMP —NH2 DMS —NH2 DPDPB —SH disulphide linker DSG —NH2 DSP —NH2 disulphide linker DSS —NH2 DST —NH2 sulfo-DST —NH2 water-soluble DTBP —NH2 disulphide linker DTSSP —NH2 disulphide linker EGS —NH2 sulfo-EGS —NH2 water-soluble SPBP —NH2 - Biotinylation Agents
Agent Reactivity Comments biotin-BMCC —SH biotin-DPPE* preparation of biotinylated liposomes biotin-LC-DPPE* preparation of biotinylated liposomes biotin-HPDP —SH disulphide linker biotin-hydrazide carbohydrate biotin-LC-hydrazide carbohydrate iodoacetyl-LC-biotin —NH2 NHS-iminobiotin —NH2 reduced affinity for avidin NHS-SS-biotin —NH2 disulphide linker photoactivatable biotin nucleic acids sulfo-NHS-biotin —NH2 water-soluble sulfo-NHS-LC-biotin —NH2
Notes:
DPPE = dipalmitoylphosphatidylethanolamine;
LC = long chain
- Agents for Protein Modification
Agent Reactivity Function Ellman's reagent —SH quantifies/detects/protects DTT —S.S— reduction 2-mercaptoethanol —S.S— reduction 2-mercaptylamine —S.S— reduction Traut's reagent —NH2 introduces —SH SATA —NH2 introduces protected —SH AMCA-NHS —NH2 fluorescent labelling AMCA-hydrazide carbohydrate fluorescent labelling AMCA-HPDP —S.S— fluorescent labelling SBF-chloride —S.S— fluorescent detection of —SH N-ethylmaleimide —S.S— blocks —SH NHS-acetate —NH2 blocks and acetylates —NH2 citraconic anhydride —NH2 reversibly blocks and introduces negative charges DTPA —NH2 introduces chelator BNPS-skatole tryptophan cleaves tryptophan residue Bolton-Hunter —NH2 introduces iodinable group - Linking agents used in accordance with the invention will in general bring about linking of vector to reporter or reporter to reporter with some degree of specificity, and may also be used to attach one or more therapeutically active agents.
- Ultrasound imaging modalities which may be used in accordance with the invention include two- and three-dimensional imaging techniques such as B-mode imaging (for example using the time-varying amplitude of the signal envelope generated from the fundamental frequency of the emitted ultrasound pulse, from sub-harmonics or higher harmonics thereof or from sum or difference frequencies derived from the emitted pulse and such harmonics, images generated from the fundamental frequency or the second harmonic thereof being preferred), colour Doppler imaging and Doppler amplitude imaging, and combinations of the two latter with any of the modalities (techniques) above. Surprisingly, the second harmonic signals from the targeted monolayer microspheres were found to be excellent when used in accordance with the present invention. To reduce the effects of movement, successive images of tissues such as the heart or kidney may be collected with the aid of suitable synchronisation techniques (e.g. gating to the ECG or respiratory movement of the subject). Measurement of changes in resonance frequency or frequency absorption which accompany arrested or retarded microbubbles may also usefully be made to detect the contrast agent.
- The present invention provides a tool for therapeutic drug delivery in combination with vector-mediated direction of the product to the desired site. By “therapeutic” or “drug” is meant an agent having a beneficial effect on a specific disease in a living human or non-human animal. Whilst combinations of drugs and ultrasound contrast agents have been proposed in, for example, WO-A-9428873 and WO-A-9507072, these products lack vectors having affinity for particular sites and thereby show comparitively poor specific retention at desired sites prior to or during drug release.
- Therapeutic compounds used in accordance with the present invention may be encapsulated in the interior of the microbubbles or attached to or incorporated in the encapsulating walls. Thus, the therapeutic compound may be linked to a part of the wall, for example through covalent or ionic bonds, or may be physically mixed into the encapsulating material, particularly if the drug has similar polarity or solubility to the membrane material, so as to prevent it from leaking out of the product before it is intended to act in the body. The release of the drug may be initiated merely by wetting contact with blood following administration or as a consequence of other internal or external influences, e.g. dissolution processes catalyzed by enzymes or the use of of ultrasound. The destruction of gas-containing microparticles using external ultrasound is a well known phenomenon in respect of ultrasound contrast agents, e.g. as described in WO-A-9325241; the rate of release may be varied depending on the type of therapeutic application, using a specific amount of ultrasound energy from the transducer.
- The therapeutic agent may be covalently linked to the encapsulating membrane surface using a suitable linking agent, e.g. as described herein. Thus, for example, one may initially prepare a phospholipid or lipopeptide derivative to which the drug is bonded through a biodegradable or selectively cleavable linker followed by incorporation of the material into the microbubble. Alternatively lipidated drug molecules which do not require processing to liberate an active drug are incorporated directly into the membrane. The active lipidated-drug can be released by increasing the strength of the ultrasound beam.
- Exemplary drug delivery systems suitable for use in the present compositions include any known therapeutic drugs or active analogues thereof containing thiol groups which are coupled to thiol containing microbubbles under oxidative conditions yielding disulphide bridges. In combination with a vector or vectors the drug/vector modified microbubbles are allowed to accumulate in the target tissue. Administration of a reducing agent such as reduced glutathione then liberates the drug molecule from the targeted microbubble in the vicinity of the target cell increasing the local concentration of the drug and enhancing therapeutic effect. The product may also be prepared without the therapeutic if desired. The drug may then be coupled to or coated on the microbubbles prior to use. Thus, for example, a therapeutic could be added to a suspension of microbubbles in aqueous media and shaken in order to attach or adhere the therapeutic to the microbubbles.
- Other drug delivery systems include vector modified phospholipid membranes doped with lipopeptide structures comprising a poly-L-lysine or poly-D-lysine chain in combination with a targeting vector. Applied to gene therapy/antisense technologies with particular emphasis on receptor-mediated drug delivery the microbubble carrier is condensed with DNA or RNA via elecrostatic interaction with the polycation. This method has the advantage that the vector or vectors used for targeted delivery are not directly attached to the polysine carrier moiety. The polylysine chain is also anchored more tightly in the microbubble membrane due to the presence of the lipid chains. The use of ultrasound to increase the effectiveness of delivery is also considered useful.
- Alternatively free polylysine chains are firstly modified with drug or vector molecules then condensed onto the negative surface of targeted microbubbles.
- Representative and non-limiting examples of drugs useful in accordance with the invention include antineoplastic agents such as vincristine, vinblastine, vindesine, busulfan, chlorambucil, spiroplatin, cisplatin, carboplatin, methotrexate, adriamycin, mitomycin, bleomycin, cytosine arabinoside, arabinosyl adenine, mercaptopurine, mitotane, procarbazine, dactinomycin (antinomycin D), daunorubicin, doxorubicin hydrochloride, taxol, plicamycin, aminoglutethimide, estramustine, flutamide, leuprolide, megestrol acetate, tamoxifen, testolactone, trilostane, amsacrine (m-AMSA), asparaginase (L-asparaginase), etoposide, interferon a-2a and 2b, blood products such as hematoporphyrins or derivatives of the foregoing; biological response modifiers such as muramylpeptides; antifungal agents such as ketoconazole, nystatin, griseofulvin, flucytosine, miconazole or amphotericin B; hormones or hormone analogues such as growth hormone, melanocyte stimulating hormone, estradiol, beclomethasone dipropionate, betamethasone, cortisone acetate, dexamethasone, flunisolide, hydrocortisone, methylprednisolone, paramethasone acetate, prednisolone, prednisone, triamcinolone or fludrocortisone acetate; vitamins such as cyanocobalamin or retinoids; enzymes such as alkaline phosphatase or manganese superoxide dismutase; antiallergic agents such as amelexanox; inhibitors of tissue factor such as monoclonal antibodies and Fab fragments thereof, synthetic peptides, nonpeptides and compounds downregulating tissue factor expression; inhibitors of platelets such as, GPIa, GPIb and GPIIb-IIIa, ADP receptors, thrombin receptors, von Willebrand factor, prostaglandins, aspirin, ticlopidin, clopigogrel and reopro; inhibitors of coagulation protein targets such as: FIIa FVa, FVIIa, FVIIIA, FIXa, tissue factor, hepatins, hirudin, hirulog, argatroban, DEGR-rFVIIa and annexin V; inhibitors of fibrin formation and promoters of fibrionolysis such as t-PA, urokinase, Plamin, Streptokinase, rt-Plasminogen Activator and rStaphylokinase; antiangiogenic factors such as medroxyprogesteron, pentosan polysulphate, suramin, taxol, thalidomide, angiostatin, interferon-alpha, metalloproteinase inhibitors, platelet factor 4, somatostatin, thromobospondin; circulatory drugs such as propranolol; metabolic potentiators such as glutathione; antituberculars such as p-aminosalicylic acid, isoniazid, capreomycin sulfate, cyclosexine, ethambutol, ethionamide, pyrazinamide, rifampin or streptomycin sulphate; antivirals such as acyclovir, amantadine, azidothymidine, ribavirin or vidarabine; blood vessel dilating agents such as diltiazem, nifedipine, verapamil, erythritol tetranitrate, isosorbide dinitrate, nitroglycerin or pentaerythritol tetranitrate; antibiotics such as dapsone, chloramphenicol, neomycin, cefaclor, cefadroxil, cephalexin, cephradine, erythromycin, clindamycin, lincomycin, amoxicillin, ampicillin, bacampicillin, carbenicillin, dicloxacillin, cyclacillin, picloxacillin, hetacillin, methicillin, nafcillin, penicillin, polymyxin or tetracycline; antiinflammatories such as diflunisal, ibuprofen, indomethacin, meclefenamate, mefenamic acid, naproxen, phenylbutazone, piroxicam, tolmetin, aspirin or salicylates; antiprotozoans such as chloroquine, metronidazole, quinine or meglumine antimonate; antirheumatics such as penicillamine; narcotics such as paregoric; opiates such as codeine, morphine or opium; cardiac glycosides such as deslaneside, digitoxin, digoxin, digitalin or digitalis; neuromuscular blockers such as atracurium mesylate, gallamine triethiodide, hexafluorenium bromide, metocurine iodide, pancuronium bromide, succinylcholine chloride, tubocurarine chloride or vecuronium bromide; sedatives such as amobarbital, amobarbital sodium, apropbarbital, butabarbital sodium, chloral hydrate, ethchlorvynol, ethinamate, flurazepam hydrochloride, glutethimide, methotrimeprazine hydrochloride, methyprylon, midazolam hydrochloride, paraldehyde, pentobarbital, secobarbital sodium, talbutal, temazepam or triazolam; local anaesthetics such as bupivacaine, chloroprocaine, etidocaine, lidocaine, mepivacaine, procaine or tetracaine; general anaesthetics such as droperidol, etomidate, fentanyl citrate with droperidol, ketamine hydrochloride, methohexital sodium or thiopental and pharmaceutically acceptable salts (e.g. acid addition salts such as the hydrochloride or hydrobromide or base salts such as sodium, calcium or magnesium salts) or derivatives (e.g. acetates) thereof. Other examples of therapeutics include genetic material such as nucleic acids, RNA, and DNA of natural or synthetic origin, including recombinant RNA and DNA. DNA encoding certain proteins may be used in the treatment of many different types of diseases. For example, tumor necrosis factor or interleukin-2 genes may be provided to treat advanced cancers; thymidine kinase genes may be provided to treat ovarian cancer or brain tumors; interleukin-2 genes may be provided to treat neuroblastoma, malignant melanoma or kidney cancer; and interleukin-4 genes may be provided to treat cancer.
- Lipophilic derivatives of drugs linked to the microbubble wall through hydrophobic interactions may exhibit therapeutic effects as part of the microbubble or after release from the microbubble, e.g. by use of ultrasound. If the drug does not possess the desired physical properties, a lipophilic group may be introduced for anchoring the drug to the membrane. Preferably the lipophilic group should be introduced in a way that does not influence the in vivo potency of the molecule, or the lipophilic group may be cleaved releasing the active drug. Lipophilic groups may be introduced by various chemical means depending on functional groups available in the drug molecule. Covalent coupling may be effected using functional groups in the drug molecule capable of reacting with appropriately functionalised lipophilic compounds. Examples of lipophilic moieties include branched and unbranched alkyl chains, cyclic compounds, aromatic residues and fused aromatic and non-aromatic cyclic systems. In some instances the lipophilic moiety will consist of a suitably functionalised steroid, like cholesterol and related compounds. Examples of functional groups particularly suitable for derivatisation include nucleophilic groups like amino, hydroxy and sulfhydryl groups. Suitable processes for lipophilic derivatisation of any drug containing a sulfhydryl group, like captopril, may include direct alkylation, e.g. reaction with an alkyl halide under basic conditions and thiol ester formation by reaction with an activated carboxylic acid. Representative examples of derivatisation of any drug having carboxylic functions, like atenolol and chlorambucil, include amide and ester formation by coupling of amines and alcohols, respectively, possesing requested physical properties. A preferred aspect is attachment of cholesterol to a therapeutic compound by forming a degradable ester bond.
- A preferred application of the present invention relates to angiogenesis, which is the formation of new blood vessels by branching from existing vessels. The primary stimulus for this process may be inadequate supply of nutrients and oxygen (hypoxia) to cells in a tissue. The cells may respond by secreting angiogenetic factors, of which there are many; one example is vascular endothelial growth factor. These factors initiate the secretion of proteolytic enzymes which break down the proteins of the basement membrane, as well as inhibitors which limit the action of these potentially harmful enzymes. The combined effect of loss of attachment and signals from the receptors for angiogenetic factors is to cause the endothelial cells to move, multiply, and rearrange themselves, and finally to synthetise a basement membrane around the new vessels.
- Tumors must initiate angiogenesis when they reach millimeter size in order to keep up their rate of growth. As angiogenesis is accompanied by characteristic changes in the endothelial cells and their environment, this process is a promising target for therapeutic intervention. The transformations accompanying angiogenesis are also very promising for diagnosis, a preferred example being malignant disease, but the concept also shows great promise in inflammation and a variety of inflammation-related diseases. These factors are also involved in re-vascularisation of infarcted parts of the myocardium, which occurs if a stenosis is released within a short time.
- A number of known receptors/targets associated with angiogenesis are given in subsequent tables. Using the targeting principles described in the present disclosure, angiogenesis mav be detected by the majority of the imaging modalities in use in medicine. Contrast-enhanced ultrasound may possess additional advantages, the contrast medium being microspheres which are restricted to the interior of blood vessels. Even if the target antigens are found on many cell types, the microspheres will attach exclusively to endothelial cells.
- So-called prodrugs may also be used in agents according to the invention. Thus drugs may be derivatised to alter their physicochemical properties and to adapt them for inclusion into the reporter; such derivatised drugs may be regarded as prodrugs and are usually inactive until cleavage of the derivatising group regenerates the active form of the drug.
- By targeting a gas-filled microbubble containing a prodrug-activating enzyme to areas of pathology one may image targeting of the enzyme, making it possible to visualise when the micobubbles are targeted properly to the area of pathology and at the same time have disappeared from non-target areas. In this way one can determine the optimal time for injection of prodrug into individual patients.
- Another alternative is to incorporate the prodrug, the prodrug-activating enzyme and the vector in the same microbubble in a system where the prodrug will only be activated after some external stimulus. Such a stimulus may, for example, be a tumour-specific protease as described above, or bursting of the bubbles by external ultrasound after the desired targeting has been achieved.
- Therapeutics may easily be delivered in accordance with the invention to diseased or necrotic areas including the heart and vasculature in general, and to the liver, spleen and kidneys and other regions such as the lymph system, body cavities or gastrointestinal system.
- Products according to the present invention may be used for targeted therapeutic delivery either in vivo or in vitro. In the latter context the products may be useful in in vitro systems such as kits for diagnosis of different diseases or characterisation of different components in blood or tissue samples. Similar techniques to those used to attach certain blood components or cells to polymer particles(e.g. monodisperse magnetic particles) in vitro to separate them from a sample may be used in the present invention,using the low density of the reporter units in agents of the present invention to effect separation of the gas-containing material by floatation and repeated washing.
- Vectors which may be usefully employed in generating multiple-specific targetable contrast agents according to the invention include the following:
- i) Antibodies, which can be used as vectors for a very wide range of targets, and which have advantageous properties such as very high specificity, high affinity (if desired), the possiblity of modifying affinity according to need etc. Whether or not antibodies will be bioactive will depend on the specific vector/target combination. Both conventional and genetically engineered antibodies may be employed, the latter permitting engineering of antibodies to particular needs, e.g. as regards affinity and specificity. The use of human antibodies may be preferred to avoid possible immune reactions against the vector molecule. A further useful class of antibodies comprises so-called bispecific antibodies, i.e. antibodies having specificity for two different target molecules in one antibody molecule. Such antibodies may, for example, be useful in promoting formation of bubble clusters and may also be used for various therapeutic purposes, e.g. for carrying toxic moieties to the target. Various aspects of bispecific antibodies are described by McGuinness, B. T. et al. in Nat. Biotechnol. (1996) 14, 1149-1154; by George, A. J. et al. in J. Immunol. (1994) 152, 1802-1811; by Bonardi et al. in Cancer Res. (1993) 53, 3015-3021; and by French, R. R. et al. in Cancer Res. (1991) 51, 2353-2361.
- ii) Cell adhesion molecules, their receptors, cytokines, growth factors, peptide hormones and pieces thereof. Such vectors rely on normal biological protein-protein interactions with target molecule receptors, and so in many cases will generate a biological response on binding with the targets and thus be bioactive; this may be a relatively insignificant concern with vectors which target proteoglycans.
- iii) Non-peptide agonists/antagonists or non-bioactive binders of receptors for cell adhesion molecules, cytokines, growth factors and peptide hormones. This category may include non-bioactive vectors which will be neither agonists nor antagonist but which may nonetheless exhibit valuable targeting ability.
- iv) Oligonucleotides and modified oligonucleotides which bind DNA or RNA through Watson-Crick or other types of base-pairing. DNA is usually only present in extracelluar space as a consequence of cell damage, so that such oligonucleotides, which will usually be non-bioactive, may be useful in, for example, targeting of necrotic regions, which are associated with many different pathological conditions. Oligonucleotides may also be designed to bind to specific DNA- or RNA-binding proteins, for example transcription factors which are very often highly overexpressed or activated in tumour cells or in activated immune or endothelial cells. Combinatorial libraries may be used to select oligonucleotides which bind specifically to possible target molecules (from proteins to caffeine) and which therefore may be employed as vectors for targeting.
- v) DNA-binding drugs may behave similarly to oligonuclotides, but may exhibit biological acitvity and/or toxic effects if taken up by cells.
- vi) Various small molecules, including bioactive compounds known to bind to biological receptors of various kinds. Such vectors or their targets may be used to generate non-bioactive compounds binding to the same targets.
- vii) Vector molecules may be selected from combinatorial libraries without necessarily knowing the exact molecular target, by functionally selecting (in vitro, ex vivo or in vivo) for molecules binding to the region/structure to be imaged.
- viii) Various small molecules, including bioactive compounds known to bind to biological receptors of various kinds. Such vectors or their targets may be used for generate non-bioactive compounds binding to the same targets.
- ix) Proteins or peptides which bind to glucosamino-glycan side chains e.g. haparan sulphate, including glucosoaminoglycan-binding portions of larger molecules, since binding to such glucosoaminoglycans side chains does not result in a biological response. Proteoglycans are not found on red blood cells, thus eliminating undesirable adsorption to these cells.
- Other peptide vectors and lipopeptides thereof of particular interest for targeted ultrasound imaging are listed below: Atherosclerotic plaque binding peptides such as YRALVDTLK, YAKFRETLEDTRDRMY and RALVDTEFKVKQEAGAK; Thrombus binding peptides such as NDGDFEEIPEEYLQ and GPRG; Platelet binding peptides such as PLYKKIIKKLLES; and cholecystokinin, α-melanocyte-stimulating hormone, heat stable enterotoxin 1, vasoactive intestinal peptide, synthetic alpha-M2 peptide from the third heavy chain complementarity-determining region and analogues thereof for tumor targeting.
- The following tables identify various receptors which may be targeted by particular types of vectors and consequent areas of use for targetable ultrasound contrast agents according to the invention which contain such vectors.
- Protein and Peptide Vectors—Antibodies
Vector type Receptor Comments/areas of use Ref antibodies CD34 vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies ICAM-1 vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies ICAM-2 vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies ICAM-3 vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies E-selectin vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies P-selectin vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies PECAM vascular diseases in general, (general) normal vessel wall (e.g myocardium), activated endothelium, immune cells antibodies Integrins, vascular diseases in general, (general) e.g. VLA-1, normal vessel wall (e.g VLA-2, VLA-3, myocardium), activated VLA-4, VLA-5, endothelium, immune cells VLA-6, β1α7, β1α8, β1αV, LFA-1, Mac-1, CD41a, etc. antibodies GlyCAM Vessel wall in lymph nodes (general) (quite specific for lymph nodes) antibodies MadCam 1 Vessel wall in lymph nodes (general) (quite specific for lymph nodes) antibodies fibrin Thrombi (general) antibodies Tissue Activated endothelium, (general) Factor tumours antibodies Myosin Necrosis, myocardial (general) infaction antibodies CEA Tumours (general) (carcinoembryonal antigen) antibodies Mucins Tumours (general) antibodies Multiple Tumours (general) drug resistance protein antibodies Prostate Prostate cancer (general) specific antigen antibodies Cathepsin Tumours (proteases of various (general) B kinds are often more or less specifically overexpressed in a variety of tumours - Cathepsin B is such a protease) antibodies Transferrin Tumors, (general) receptor vessel wall MoAb 9.2.27 Tumours Antigen upregulated on cell growth VAP-1 Adhesion molecule Band 3 Upregulated during phagocytic protein activity CD44 tumor cells β2- general microglobulin MHC class I general antibody integrin tumors, angiogenisis c αvβ3 antibodies CD44 tumour cells a antibodies β2- general b microglobulin antibodies MHC class 1 general b
a. ) Heider, K. H., M. Sproll, S. Susani, E. Patzelt, P. Beaumier, E. Ostermann, H. Ahorn, and G. R. Adolf. 1996. “Characterization of a high-affinity monoclonal antibody specific for CD44v6 as candidate for immunotherapy of squamous cell carcinomas”. Cancer Immunology Immunotherapy 43: 245-253.
b). I. Roitt, J. Brostoff, and D. Male. 1985. Immunology, London: Gower Medical Publishing, p. 4.7
c.) Stromblad, S., and D. A. Cheresh. 1996. “Integrins, angiogenesis and vascular cell survival”. Chemistry & Biology 3: 881-885.
- Protein and Peptide Vectors—Cell Adhesion Molecules etc.
Vector type Receptor Comments/areas of use Ref L-selectin CD34 vascular diseases in MadCAM1 general, normal vessel wall GlyCam 1 (e.g myocardium), activated endothelium, Lymph nodes Other selectins carbohydrate vascular diseases in ligands general, normal vessel wall (sialyl Lewis x) (e.g myocardium), activated heparan sulfate endothelium RGD-peptides integrins angiogenisis PECAM PECAM, Endothelium, and other Cells in immune system Integrins, Laminin, Endothelium, e.g. VLA-1, VLA-2, collagen, Vessel wall VLA-3, VLA-4, fibronectin, etc. VLA-5, VLA-6, VCAM-1, β1α7, β1α8, thrombospondin, β1αV, LFA-1, Mac-1, vitronectin CD41a, etc. etc. Integrin Integrins, Cells in immune system receptors, e.g. VLA-1, vessel wall e.g. Laminin, VLA-2, VLA-3, etc. collagen, VLA-4, VLA-5, fibronectin, VLA-6, β1α7, VCAM-1, β1α8, β1αV, thrombospondin, LFA-1, Mac-1, vitronectin CD41a, etc. etc. Nerve cell proteoglycans angiogenesis c adhesion N-CAM molecule (N-CAM) (homophilic) RGD-peptides integrins - Vectors Comprising Cytokines/Growth Factors/Peptide Hormones and Fragments Thereof
Vector type Receptor Comments/areas of use Ref Epidermal growth EGF-receptor or Tumours factor related receptors Nerve growth NGF-receptor Tumours factor Somatostatin ST-receptor Tumours Endothelin Endothelin- Vessel wall receptor Interleukin-1 IL-1-receptor Inflammation, activated cells of different kinds Interleukin-2 IL-2-receptor Inflammation, activated cells of different kinds Chemokines (ca. Chemokine Inflammation 20 different receptors, cytokines partly proteoglycans sharing receptors) Tumour necrosis TNF-receptors Inflammation factor Parathyroid PTH-receptors Bone diseases hormone Kidney diseases Bone BMP-receptors Bone Diseases Morphogenetic Protein Calcitonin CT-receptors Bone diseases Colony Corresponding Endothelium stimulating specific factors (G-CSF, receptors, GM-CSF, M-CSF, proteoglycans IL-3) Insulin like IGF-I receptor Tumours, growth factor I other growing tissues Atrial ANF-receptors Kidney, Natriuretic vessel wall Factor Vasopressin Vasopressin Kidney, receptor vessel wall VEGF VEGF-receptor Endothelium, regions of angiogenesis Fibroblast FGF-receptors, Endothelium growth factors Proteoglycans Angiogenesis Schwann cell proteoglycans growth factor specific receptors - Miscellaneous Protein and Peptide Vectors
Vector type Receptor Comments/areas of use Ref Streptavidin Kidney Kidney diseases Bacterial Fibronectin Vessel wall fibronectin- binding proteins Fc-part of Fc-receptors Monocytes antibodies macrophages liver Transferrin transferrin- Tumours receptor vessel walls Streptokinase/ thrombi thrombi tissue plasminogen activator Plasminogen, Fibrin Thrombi, plasmin tumours Mast cell proteoglycans proteinases Elastase proteoglycans Lipoprotein proteoglycans lipase Coagulation proteoglycans enzymes Extracellular proteoglycans superoxide dismutase Heparin cofactor proteoglycans II Retinal survival proteoglycans factor specific receptors Heparin-binding proteoglycans brain mitogen specific receptors Apolipoprotein, proteoglycans e.g. specific apolipoprotein B receptors (e.g., LDL receptor) Apolipoprotein E LDL receptor proteoglycans Adhesion- proteoglycans promoting proteins, e.g. Purpurin Viral coat proteoglycans proteins, e.g. HIV, Herpes Microbial “Antigen 85” fibronectin, collagen, adhesins complex of fibrinogen, vitronectin, mycobacteria heparan sulfate β-amyloid proteoglycans β-amyloid accumulates in precursor Alzheimer's disease Tenascin, heparan sulfate, e.g. tenascin C integrins - Vectors Comprising Non-peptide Agonists/Antagonists of Cytokines/Growth Factors/Peptide Hormones/Cell Adhesion Molecules
Vector type Receptor Comments/areas of use Ref Endothelin Endothelin Vessel wall antagonist receptor Desmopressin Vasopressin Kidney (vasopressin receptor Vessel wall analogue) Demoxytocin Oxytocin Reproductive organs, (oxytocin Receptor Mammary glands, analogue) Brain Angiotensin II Angiotensin II Vessel wall receptor receptors brain antagonists adrenal gland CV-11974, TCV-116 non-peptide RGD- integrins Cells in immune system analogues vessel wall etc. - Vectors Comprising Anti-angiogenic Factors
Vector type Target Comments/areas of use Ref Angiostatin EC of tumors plasminogen fragment K cartilage-derived EC of tumors J inhibitor β-Cyclodextrin tumors, C tetradecasulfate inflammation fumagillin and analogs tumors, E inflammation Interferon-α EC of tumors K Interferon-γ EC of tumors E interleukin-12 EC of tumors E linomide tumors, A inflammation medroxyprogesterone EC of tumors K metalloproteinase EC of tumors K inhibitors pentosan polysulfate EC of tumors K platelet factor 4 EC of tumors M Somatostatin EC of tumors K Suramin EC of tumors K Taxol EC of tumors K thalidomide EC of tumors K Thrombospondin EC of tumors K - Vectors Comprising Angiogenic Factors
Comments/areas Vector type Target of use Ref acidic fibroblast EC of tumors K growth factor adenosine EC of tumors K Angiogenin EC of tumors K Angiotensin II EC of tumors K basement membrane tumors e.g., tenascin, M components collagen IV basic fibroblast EC of tumors K growth factor Bradykinin EC of tumors K Calcitonin gene- EC of tumors K related peptide epidermal growth EC of tumors K factor Fibrin tumors K Fibrinogen tumors K Heparin EC of tumors K histamine EC of tumors K hyaluronic acid EC of tumors K or fragments thereof Interleukin-1α EC of tumors K laminin, laminin EC of tumors K fragments nicotinamide EC of tumors K platelet acti- EC of tumors K vating factor Platelet-derived EC of tumors K endothelial growth factor prostaglandins EC of tumors K E1, E2 spermine EC of tumors K spermine EC of tumors K Substance P EC of tumors K transforming EC of tumors K growth factor-α transforming EC of tumors K growth factor-β Tumor necrosis factor-α EC of tumors K vascular endo- EC of tumors K thelial growth factor/vascular permeability factor vitronectin A - Vector Molecules Other than Recognized Angiogenetic Factors with Known Affinity for Receptors Associated with Angiogenesis
Vector type Target Comments/areas of use Ref angiopoietin tumors, B inflammation α2-antiplasmin tumors, inflammation combinatorial tumors, for instance: compounds libraries, compounds inflammation that bind to basement from membrane after degradation endoglin tumors, D inflammation endosialin tumors, D inflammation endostatin [collagen tumors, M fragment] inflammation Factor VII related tumors, D antigen inflammation fibrinopeptides tumors, ZC inflammation fibroblast growth tumors, E factor, basic inflammation hepatocyte growth tumors, I factor inflammation insulin-like growth tumors, R factor inflammation interleukins tumors, e.g.,: IL-8 I inflammation leukemia inhibitory tumors, A factor inflammation metalloproteinase tumors, e.g., batimastat E inhibitors inflammation Monoclonal antibodies tumors, for instance: to inflammation angiogenetic factors or their receptors, or to components of the fibrinolytic system peptides, for instance tumors, B, Q cyclic RGDDFV inflammation placental growth factor tumors, J inflammation placental tumors, E proliferin-related inflammation protein plasminogen tumors, M inflammation plasminogen activators tumors, D inflammation plasminogen activator tumors, U, V inhibitors inflammation platelet activating tumors, inhibitors of angiogenesis A factor antagonists inflammation platelet-derived growth tumors, E factor inflammation pleiotropin tumors, ZA inflammation proliferin tumors, E inflammation proliferin related tumors, E protein inflammation selectins tumors, e.g., E-selectin D inflammation SPARC tumors, M inflammation snake venoms tumors, Q (RGD-containing) inflammation Tissue inhibitor of tumors, eg,, TIMP-2 U metalloproteinases inflammation thrombin tumors, H inflammation thrombin-receptor-activating tumors, H tetradecapeptide inflammation thymidine phosphorylase tumors, D inflammation tumor growth factor tumors, ZA inflammation - Receptors/Targets Associated with Angiogenesis
Vector type Target Comments/areas of use Ref biglycan tumors, dermatan sulfate X inflammation proteoglycan CD34 tumors, L inflammation CD44 tumors, F inflammation collagen type I, IV, tumors, A VI, VIII inflammation decorin tumors, dermatan sulfate Y inflammation proteoglycan dermatan sulfate tumors, X proteoglycans inflammation endothelin tumors, G inflammation endothelin receptors tumors, G inflammation fibronectin tumors P Flk-1/KDR, Flt-4 tumors, VEGF receptor D inflammation FLT-1 (fms-like tumors, VEGF-A receptor O tyrosine kinase) inflammation heparan sulfate tumors, P inflammation hepatocyte growth tumors, I factor receptor (c-met) inflammation insulin-like growth tumors, R factor/mannose-6- inflammation phosphate receptor integrins: Tumors, D, β3 and β5, inflammation P integrin αvβ3, integrin α6β1, laminin receptor integrins α6, integrins β1, integrin α2β1, integrin αvβ3, integrin α5 subunit of the fibronectin receptor integrin αVβ5, fibrin receptors. Intercellular adhesion tumors, P molecule-1 and -2 inflammation Jagged gene product tumors, T inflammation Ly-6 tumors, a lymphocyte activation N inflammation protein matrix tumors, D metalloproteinases inflammation MHC class II tumors, inflammation Notch gene product tumors, T inflammation Osteopontin tumors Z PECAM tumors, alias CD31 P inflammation plasminogen activator tumors, ZC receptor inflammation platelet-derived growth tumors, E factor receptors inflammation Selectins: E-, P- tumors, D inflammation Sialyl Lewis-X tumors, blood group antigen M inflammation stress proteins: tumors, molecular chaperones glucose regulated, heat inflammation shock families and others syndecan tumors, T inflammation thrombospondin tumors, M inflammation TIE receptors tumors, tyrosine kinases with Ig- E inflammation and EGF-Iike domains tissue factor tumors, Z inflammation tissue inhibitor of tumors, e.g., TIMP-2 U metalloproteinases inflammation transforming growth tumors, E factor receptor inflammation urokinase-type tumors, D plasminogen activator inflammation receptor Vascular cellular tumors, D adhesion molecule inflammation (VCAM) Vascular endothelial tumors, growth factor related inflammation protein Vascular endothelial tumors, K growth factor-A inflammation receptor von Willebrand factor- tumors, L related antigen inflammation - Oligonucleotide Vectors
Vector type Receptor Comments/areas of use Ref Oligonucleotides DNA made Tumours complementary to available by Myocardial infarction repeated necrosis All other diseases that sequences, e.g. involves necrosis genes for ribosomal RNA, Alu-sequences Oligonucleotides DNA made Tumours complementary to available by disease-specific necrosis in a mutations (e.g. region of the mutated relevant disease oncogenes). Oligonucleotides DNA of infective Viral or bacterial complementary to agent infections DNA of infecting agent. Triple or As in above As in above examples quadruple-helix examples forming Oligonucleotides Oligonucleotides DNA-binding Tumours with recognition protein, e.g. Activated endothelium sequence for transcription Activated immune cells DNA-or RNA- factors (often binding proteins overexpressed/ activated in tumours or activated endothelium/ immune cells - Modified Oligonucleotide Vectors
Vector type Receptor Comments/areas of use Ref Phosphorothioate As for As for unmodified oligos oligos unmodified oligos 2′-O-methyl As for As for unmodified oligos substituted unmodified oligos oligos circular oligos As for As for unmodified oligos unmodified oligos oligos As for As for unmodified oligos containing unmodified hairpin oligos structure to decrease degradation oligos with As for As for unmodified oligos terminal unmodified phosphorothioate oligos 2′-fluoro oligos As for As for unmodified oligos unmodified oligos 2′-amino oligos As for As for unmodified oligos unmodified oligos DNA-binding As for Increased binding affinity drugs conjugated unmodified as compared to pure oligos to oligos (for oligos examples, see below) Peptide Nucleic As for Increased binding affinity Acids (PNAs, unmodified and stability compared to oligonucleotidss oligos standard oligos. with a peptide backbone) - Nucleoside and Nucleotide Vectors
Vector type Receptor Comments/areas of use Ref Adenosine or Adenosine Vessel wall analogues receptors Heart ADP, UDP, UTP Various Many tissues, e.g. brain, and others nucleotide spinal cord, kidney, spleen receptors - Receptors Comprising DNA-binding Drugs
Vector type Receptor Comments/areas of use Ref acridine DNA made Tumours, derivatives available by Myocardial infarction and distamycin necrosis all other diseases involving netropsin necrosis or other processes actinomycin D liberating DNA from cells echinomycin bleomycin etc. - Receptors Comprising Protease Substrates
Vector type Receptor Comments/areas of use Ref Peptidic or non- Cathepsin B Tumours, a variety of which peptidic may more or less specifically substrates overexpress proteases of various kinds, e.g. Cathepsin B - Receptors Comprising Protease Inhibitors
Vector type Receptor Comments/areas of use Ref Peptidic or non- Cathepsin B Tumours, a variety of which peptidic may more or less specifically inhibitors overexpress proteases of e.g. N-acetyl- various kinds, e.g. Leu-Leu- Cathepsin B norleucinal bestatin Aminopeptidases Tumours, ([(2S,3R)-3- e.g. on cell surfaces Amino-2-hydroxy- 4-phenyl- butanoyl]-L- leucine hydrochloride) Pefabloc (4-(2- Serine proteases Tumours, aminoethyl)- vessel wall benzenesulfonyl etc. fluoride hydrochloride) Commercially Angiotensin Endothelial cells available converting inhibitors enzyme e.g. kaptopril enalapril ricionopril Low specificity Coagulation Vessel wall injury, non-peptidic factors tumours, compounds etc. Protease nexins proteoglycans (extracellular protease inhibitors) Antithrombin proteoglycans, Coagulation actors - Vectors from Combinatorial Libraries
Vector type Receptor Comments/areas of use Ref Antibodies with Any of above Any diseased or normal structure targets - or may structure of interest, e.g. determined be unknown when thrombi, tumours or walls of during make functional myocardial vessels generation selection of process vector binding to chosen diseased structure Peptides with Any of above Any diseased or normal sequence targets - or may structure of interest, e.g. determined be unknown when thrombi, tumours or walls of during make functional myocardial vessels generation selection of process vector binding to chosen diseased structure Oligonucleotides Any of above Any diseased or normal with sequence targets - or may structure of interest, e.g. determined be unknown when thrombi, tumours or walls of during make functional myocardial vessels generation selection of process vector binding to chosen diseased structure Modifications of Any of above Any diseased or normal oligos obtained targets - or may structure of interest, e.g. as above be unknown when thrombi, tumours or walls of make functional myocardial vessels selection of vector binding to chosen diseased structure Other chemicals Any of above Any diseased or normal with structure targets - or may structure of interest, e.g. determined be unknown when thrombi, tumours or walls of during make functional myocardial vessels generation selection of process vector binding to chosen diseased structure - Carbohydrate Vectors
Vector type Receptor Comments/areas of use Ref neo- macrophages general activation/ glycoproteins inflammation oligosaccharides Asialo- liver with terminal glycoprotein galactose receptor Hyaluronan aggrecan (a proteoglycan) “link proteins” cell-surface receptors: CD44 Mannose Blood brain barrier, Brain tumours and other diseases causing changes in BBB Bacterial Blood brain barrier, glycopeptides Brain tumours and other diseases causing changes in BBB - Lipid Vectors
Vector type Receptor Comments/areas of use Ref LDL-like lipids LDL-receptor Atherosclerosis - Small Molecule Vectors
Vector type Receptor Comments/areas of use Ref Adrenalin Corresponding receptors Betablockers Adrenergic beta- Myocardium for beta-1 receptors blockers Alpha-blockers Adrenergic Vessel wall alpha-receptors benzodiazepines serotonin- Serotonin- analogues receptors anti-histamines Histamine- Vessel wall receptors Acetyl-choline ACh-receptors receptor antagonists verapamil Ca2+-channel Heart muscle blocker nifedipin Ca2+-channel Heart muscle blocker Amiloride Na+/H+-exchanger Blocks this exchanges in kidney and is generally upregulated in cells stimulated by growth factors. Digitalis Na+/K+-ATP-ases myocardium glycosides peripheral vasculature, central nervous system Thromboxae/ Thromboxane/ Vessel wall, Prostaglandin prostaglandin Endothelium receptor receptors antagonists or agonists Glutathione Glutathione- Lung, receptors Brain Leukotriene- receptors Biotin biotin transport protein on cell surface Folate folate transport Tumours protein on cell surface Riboflavin riboflavin transport protein on cell surface Methotrexate folate transport protein on cell surface chlorambucil general transport mechanisms
References to the Preceding Tables - A. Auerbach, W., and R. Auerbach. 1994. “Angiogenesis inhibition: a review”. Pharmac. Ther. 63: 265-311.
- B. Barinaga, M. 1997. “Designing Therapies That Target Tumor Blood Vessels”. Science 275 (January 24): 482-484.
- C. Folkman, J., P. B. Weisz, M. M. Joullie, W. W. Li, and W. R. Ewing. 1989. “Control of Angiogenesis With Synthetic Heparin Substitutes”. Science 243: 1490-1493.
- D. Fox, S. B., and A. L. Harris. 1997. “Markers of tumor angiogenesis: Clinical applications in prognosis and anti-angiogenic therapy”. Investigational New Drugs 15 (1): 15-28.
- E. Gastl, G., T. Hermann, M. Steurer, J. Zmija, E. Gunsilius, C. Unger, and A. Kraft. May 1997. “Angiogenesis as a target for tumor treatment”. Oncology 54 (3): 177-184.
- F. Griffioen, A. W., M. J. H. Coenen, C. A. Damen, S. M. M. Hellwig, D. H. J. Vanweering, W. Vooys, G. H. Blijham, and G. Groenewegen. 1 Aug. 1997. “CD44 is involved in tumor angiogenesis; an activation antigen on human endothelial cells”. Blood 90 (3): 1150-1159.
- G. Hlatky, L., P. Hahnfeldt, and C. N. Coleman. 1996. “Vacular endothelial growth factor: environmental controls and effects in angiogenesis”. Brit. J. Cancer 74 (Suppl. XXVII): S151-S156.
- H. Maragoudakis, M. E., E. Pipili-Synethos, E. Sakkoula, D. Panagiotopoulos, N. Craniti, and J. M. Matsoukas. 1996. “Inhibition of TRAP-induced angiogenesis by the tripeptide Phe-Pro-Arg, a thrombin-receptor-derived peptide analogue”. Letters in Peptide Science 3: 227-232.
- I. Nguyen, M. 1997. “Angiogenic factors as tumor markers”. Investigational New Drugs 15 (1): 29-37.
- J. Ono, M., H. Izumi, S. Yoshida, D. Gtot, S. Jimi, N. Kawahara, T. Shono, S. Ushiro, M. Ryuto, K. Kohno, Y. Sato, and M. Kuwano. 1996. “Angiogenesis as a new target for cancer treatment”. Cancer Chemoter. Pharmacol. 38 (Suppl.): S78-S82.
- K. Passe, T. J., D. A. Bluemke, and S. S. Siegelman. June 1997. “Tumor angiogenesis: Tutorial on implications for imaging”. Radiology 203 (3): 593-600.
- L. Saclarides, T. J. February 1997. “Angiogenesis in colorectal cancer”. Surgical Clinics of North America 77 (1): 253.
- M. Sage, E. H. May 1997. “Pieces of eight: Bioactive fragments of extracellular proteins as regulators of angiogenesis”. Trends in Cell Biology 7 (5): 182-186.
- N. Sagi-Assif, O., A. Traister, B. Z. Katz, R. Anavi,
- M. Eskenazy, and I. P. Witz. 1996. “TNFα and anti-Fas antibodies regulate Ly-6E.1 expression by tumor cells: A possible link between angiogenesis and Ly-6E.1”. Immunology Letters 54: 207-213.
- O. Strawn, L. M., G. McMahon, H. App, R. Schreck, W. R. Kuchler, M. P. Longhi, T. H. Hui, C. Tang, A. Levitzki, A. Gazit, I. Chen, G. Keri, L. Orfi, W. Risau, I. Flamme, A. Ullirch, K. P. Hirth, and L. K. Shawyer. 1996. “Flk-1 as a Target for Tumor Growth Inhibition”. Cancer Res. 56: 3340-3545.
- P. Stromblad, S., and D. A. Cheresh. December 1996. “Cell adhesion and angiogenesis”. Trends in Cell Biology 6 (12): 462-468.
- Q. Stromblad, S., and D. A. Cheresh. November 1996. “Integrins, angiogenesis and vascular cell survival”. Chemistry & Biology 3 (11): 881-885.
- R. Volpert, O., D. Jackson, N. Bouck, and D. I. H. Linzer. September 1996. “The insulin-like growth factor II/mannose 6-phosphate receptor is required for proliferin-induced angiogenesis”. Endocrinology 137 (9): 3871-3876.
- S. Yoshida, O. M., T. Shono, H. Izumi, T. Ishibashi, H. Suzuki, and M. Kuwano. 1997. “Involvement of Interleukin-8, Vascular Endothelial Growth Factor, and Basic Fibroblast Growth Factor in Tumor Necrosis Factor Alpha-Dependent Angiogenesis”. Mol. Cell. Biol. 17: 4015-4023.
- T. Zimrin, A. B., M. S. Pepper, G. A. McMahon, F. Nguyen, R. Montesano, and T. Maciag. 1996. “An Antisense Oligonucleotide to the Notch Ligand Jagged Enhances Fibroblast Growth Factor-induced Angiogenesis <in vitro>”. J. Biol. Chem. 271 (December 20): 32499-3502.
- U. Albini, A., R. Soldi, D. Giunciuglio, E. Giraudo, R. Benelli, R. Primo, D. Noonan, M. Salio, G. Camussi, W. Rockl, and F. Bussolino. 1996. “The angiogenesis induced by HIV-1 Tat protein is mediated by the Flk-1/KDR receptor on vacular endothelial cells”. Nature Medicine 2 (12 (December)): 1371-1374.
- V. Ferrara, N. 1996. “The biology of vascular endothelial growth factor”. in Molecular, Cellular and Clinical Aspects of Angiogenesis, ed. M. E. Maragoudakis. New York: Plenum Press.
- X. Jackson, R. L., S. J. busch, and A. J. Cardin. 1991. “Glycosaminoglycans: Molecular Properties, Protein Interactions, and Role in Physiological Processes”. Physiological Reviews 71 (2): 481-435.
- Y. Kinsella, M. G., C. K. Tsoi, H. T. Jarvelainen, and T. N. Wight. 1997. “Selective expression and processing of biglycan during migration of bovine aortic endothelial cells—The role of endogenous basic fibroblast growth factor”. Journal of Biological Chemistry 272: 318-325.
- Z. Folkman, J. 1996. Tumor angiogenesis and tissue factor. Nature Medicine 2, 167-8
- ZA. Relf, M., S. LeJeune, P. A. Scott, S. Fox, K. Smith, R. Leek, A. Moghaddam, R. Whitehouse, R. Bicknell and A: L. Harris. 1997. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor and pleiotrophin in human primary breast cancer and its relation to angiogenesis
 . Cancer Res. 57, 963-9.
. Cancer Res. 57, 963-9. - ZB. Carmeliet, P., L. Moons, M. Dewerchin, N. Mackman, T. Luther, G. Breier, V. Ploplis, M. Muller, A. Nagy, E. Plow, R. Gerard, T. Edgington, W. Risau, D. Collen. 1997. Ann, N.Y. Acad. Sci. 811, 191-206.
- ZC. Van Hinsbergh, P. Koolwijk, R. Haanemaijer. 1997. “Role of fibrin and plasminogen activators in repair-associated angiogenesis: in vitro studies with human endothelial cells” EXS 79, 391-411.
- Passe, T. J., D. A. Bluemke and S. S. Siegelman. 1997. Radiology 203: 593-600.
- Representative examples of drugs useful in accordance with the invention include: abamectin, abundiazole, acaprazine, acabrose, acebrochol, aceburic acid, acebutolol, acecainide, acecarbromal, aceclidine, ac clof nac, acedapsone, acediasulfone, acedoben, acefluranol, acefurtiamine, acefylline clofibrol, acefylline piperazine, aceglatone, aceglutamide, aceglutamide aluminium, acemetacin, acenocoumarol, aceperone, acepromazine, aceprometazine, acequinoline, acesulfame, acetaminophen, acetaminosalol, acetanilide, acetarsone, acetazolamide, acetergamine, acetiamine, acetiromate, acetohexamide, acetohydroxamic acid, acetomeroctol, acetophenazine, acetorphine, acetosulfone, acetriozate, acetryptine, acetylcholine chloride, acetylcolchinol, acetylcysteine, acetyldigitoxin, acetylleucine, acetylsalicyclic acid, acevaltrate, acexamic acid, acifran, acipimox, acitemate, acitretin, acivicin, aclantate, aclarubicin, aclatonium napadisilate, acodazole, aconiazide, aconitine, acoxatrine, acridorex, acrihellin, acrisorcin, acrivastine, acrocinide, acronine, actinoquinol, actodigin, acyclovir, adafenoxate, adamexine, ademetionine, adenosine phosphate, adibendan, adicillin, adimolol, adinazolam, adiphenine, aditeren, aditoprim, adrafinil, adrenalone, afloqualone, afurolol, aganodine, ajmaline, aklomide, alacepril, alafosfalin, alanine mustard, alanosine, alaproclate, alazanine triclofenate, albendazole, albendazole oxide, albuterol, albutoin, alclofenac, alcometasone dipropionate, alcloxa, alcuronium chloride, aldioxa, aldosterone, alepride, aletamine, alexidine, alfacalcidol, alfadex, alfadolone, alfaprostol, alfaxalone, alfentanil, alfuzosin, algestone acetonide, algestone acetophenide, alibendol, aliconazole, alifedrine, aliflurane, alimadol, alinidine, alipamide, alitame, alizapride, allantoin, alletorphine, allobarbital, alloclamide, allocupreide, allomethadione, allopurinol, allylestrenol, allyl isothicyanate, allylprodine, allylthiourea, almadrate sulfate, almasilate, almecillin, almestrone, alminoprofen, almitrine, almoxatone, alonacic, alonimid, aloxistatin, alozafone, alpertine, alphacetylmethadol, alphameprodine, alphamethadol, alphaprodine, alpha-vinylaziridinoethyl acetate, alpidem, alpiropride, alprazolam, alprenolol, alprostadil, alrestatin, altanserin, altapizone, alteconazole, althiazide, altrenogest, altretamine, aluminium acetate, aluminium clofibrate, aluminium subacetate, alverine, amadinone acetate, amafolone, amanozine, amantadine, amantanium bromide, amantocillin, ambasilide, ambazone, ambenonium chloride, ambenoxan, ambroxol, ambruticin, ambucaine, ambucetamide, ambuphylline, ambuside, ambutonium bromide, amcinafal, amcinafide, amcinonide, amdinocillin, amdinocillin pivoxil, amebucort, amedalin, ametantrone, amezepine, amezinium metilsulfate, amfenac, amfepentorex, amfetaminil, amflutizole, amfonelic acid, amicarbalide, amicibone, amicloral, amicycline, amidantel, amidapsone, amidephrine, amiflamine, amifloverine, amifloxacin, amifostine, amikacin, amikhelline, amiloride, aminacrane, amindocate, amineptine, aminobenzoic acid, aminocaproic acid, aminoethyl nitrate, aminoglutethimide, aminohippuric acid, aminometradine, aminopentamide, aminophylline, aminopromazine, aminopterin, aminopyrine, aminoquinol, aminoquinuride, aminorex, aminosalicyclic acid, aminothiadiazole, aminothiazole, amiodarone, amiperone, amipheazole, amipizone, amiprilose, amiquinsin, amisometradine, amisulpride, amiterol, amithiozone, amitraz, amitriptyline, amitriptylinoxide, amixetrine, amlexanox, amlodipine, amobarbital, amodiaquine, amogastrin, amolanone, amonofide, amoproxan, amopyroquin, amorolfine, amocanate, amosulalol, amotriphene, amoxapine, amoxecaine, amoxicillin, amoxydramine camsilate, amperozide, amphecloral, amphenidone, amphetamine, amphotalide, amphotericin B, ampicillin, ampiroxicam, amprolium, ampyrimine, ampyzine, amquinate, amrinone, amsacrine, amygdalin, amylene, amylmetacresol, amyl nitrite, anagestone acetate, anagrelide, anaxirone, anazocine, anazolene, ancarolol, ancitabine, androstanediol, androstanol propionate, androstenetrione, androstenonol propionate, anethole, anguidine, anidoxime, anilamate, anileridine, aniline, anilopam, anipamil, aniracetam, anirolac, anisacril, anisindione, anisopirol, anisoylbromacrylic acid, anitrazafen, anpirtoline, ansoxetine, antafenite, antazoline, antazonite, anthelmycin, anthiolimine, anthralin, anthramycin, antienite, antimony potassium tartrate, antimony thioglycollate, antipyrine, antrafenine, apalcillin, apazone, apicycline, apomorphine, apovincamine, apraclonidine, apramycin, aprindine, aprobarbital, aprofene, aptazapine, aptocaine, arabinosylmercaptopurine, aranotin, arbaprostil, arbekacin, arclofenin, arfendazam, arginine, arginine glutamat, arildone, arnolol, aronixil, arotinolol, arpinocid, arpromidine, arsanilic acid, arsthinol, artemisinin, articaine, asaley, ascorbic acid, ascorbyl palmitate, asocainol, aspartame, aspartic acid, asperlin, aspoxicillin, astemizole, atamestane, atenolol, atipamezole, atiprosin, atolide, atracurium besilate, atromepine, atropine, atropine oxide, auranofin, aurothoiglucose, aurothioglycanide, avilamycin-A, avridine, axamozide, azabon, azabuperone, azacitodine, azaclorzine, azaconazole, azacosterol, azacyclonol, azaftozine, azaguanidine, azaloxan, azamethonium bromide, azamulin, azanator, azanidazole, azaperone, azapicyl, azaprocin, azaquinzole, azaribine, azarole, azaserine, azaspirium chloride, azastene, azastrptonigrin, azatodine, azathioprine, azauridine, azelastine, azepexole, azepindole, azetepa, azidamfenicol, azidocillin, azimexon, azintamide, azipramine, azithromycin, azlocillin, azolimine, azosemide, azotomycin, aztreonam, azumolene, bacampicillin, baclofen, bacmecillinam, balsalazide, bamaluzole, bambuterol, bamethan, bamifylline, bamipine, bamnidazole, baquiloprim, barbexaclone, barbital, barucainide, batilol, bazinaprine, becanthone, beclamide, beclobrate, beclomethasone dipropionate, beclotiamine, befiperide, befunolol, befuraline, bekanamycin, belarizine, beloxamide, bemarinone, bemegride, bemetizide, bemitradine, benactyzine, benafentrine, benanserin, benapryzine, benaxibine, benazepril, bencianol, bencisteine, benclonidine, bencyclane, bendamustine, bendazac, bendazol, benderizine, bendroflumethiazide, benethamide penicillin, benexate, benflorex, benfosformin, benfotiamine, benfurodil hemisuccinate, benhepazone, benidipine, benmoxin, benolizime, benorilate, benorterone, benoxafos, benoxaprofen, benoxinate, benperidol, benproperine, benrixate, bensalan, benserazide, bensuldazic acid, bentazepam, bentemazole, bentiamine, bentipimine, bentiromide, benurestat, benzaldehyde, benzalkonium chloride, benzaprinoxide, benzarone, benzbromarone, benzestrol, benzethidine, benzethonium chloride, benzetimide, benzilonium, bromide, benzindopyrine, benziodarone, benzmalecene, benznidazole, benzobarbital, benzocaine, benzoclidine, benzoctamide, benzodepa, benzododecinium chloride, benzoic acid, benzoin, benzonatate, benzopyrronium bromide, benzoquinium chloride, benzotript, benzoxiquine, benzoxonium chloride, benzoyl peroxide, benzoylpas, benzphetamine, benzpiperylon, benzpyrinium bromide, benzquercin, benzquinamide, benzthiazide, benztropine, benzydamine, benzylpenicillin, benzylsulfamide, beperidium iodide, bephenium naphtoate, bepiastine, bepridil, beraprost, berberine sulfate, bermastine, bermoprofen, berythromycin, besulpamide, beslunide, beta carotene, betacetylmethadol, betahistine, betaine, betameprodine, betamethadol, betamethasone, betamethasone acetate, betamethasone acibutate, betamethasone benzoate, betamethasone dipropionate, betamethasone phosphate, betamethasone valerate, betamicin, betaprodine, betaxolol, betazole, bethanechol chloride, bethanidine, betiatide, betoxycaine, bevantolol, bevonium metilsulfate, bezafibrate, bezitramide, bialamicol, bibenzonium bromide, bibrocathol, bicifadine, biclodil, biclofibrate, biclotymol, bicozamycin, bidimazium iodine, bietamiverine, bietaserpine, bifemelane, bifepramide, bifluranol, bifonazole, binedaline, binfloxacin, binfibrate, bioallethrin, bioresmethrin, biotin, bipenamol, biperiden, biphenamine, biriperone, bisacodyl, bisantrene, bis(aziridinyl) butanediol, bisbendazole, bisbentiamine, bisfenazone, bisfentidine, bismuth betanaphthol, bismuth-triglycollamate, bismuth subgallate, bismuth subsalicylate, bisorbin, bisoprolol, bisorcic, bioxatin acetate, bispyrithione magsulfex, bithionol, bithionoloxide, bitipazone, bitoterol, bitoscantate, bleomycin, bluensomycin, bofumustine, bolandiol dipropionate, bolasterone, bolazine, boldenone undecylenate, bolenol, bolmantalate, bometolol, bopindolol, bornaprine, bornaprolol, bornelone, botiacrine, boxidine, brallobarbital, brazergoline, brefonalol, bremazocine, brequinar, bretylium tosylate, brindoxime, brivundine, brobactam, broclepride, brocresine, brocrinat, brodimoprim, brofaromine, brofezil, brofoxine, brolaconazole, brolamfetamine, bromacrylide, bromadoline, bromamid, bromazepam, bromchlorenone, bromebric acid, bromerguride, brometenamine, bromfenac, bromhexine, bromindione, bromisovalum, bromociclen, bromocriptine, bromodiphenhydramine, bromofenofos, bromopride, bromoxandide, bromperidol, bromperidol decanoate, brompheniramine, bronopol, broparestrol, broperamole, bropirimine, broquinaldol, brosotamide, brosuximide, brotianide, brotizolam, brovanexine, brovincamine, broxaldine, broxaterol, broxitalamic acid, broxuridine, broxyquinoline, bruceantin, brucine, bucainide, bucetin, buciclovir, bucillamine, bucindolol, bucladesine, buclizine, buclosamide, bucloxic acid, bucolome, bucricaine, bucromarone, bucrylate, bucumolol, budesonide, budipin, budotitane, budralazine, bufenadrine, bufeniode, bufetolol, bufexamac, bufezolac, buflomedil, bufogenin, buformin, bufrolin, bufuralol, bumadizone, bumecaine, bumepidil, bumetanide, bumetrizole, bunaftine, bunamidine, bunamiodyl, bunaprolast, bunazosin, bunitrolol, bunolol, buparvaquone, bupicomide, bupivacaine, bupranolol, buprenorphine, bupropion, buquineran, buquinolate, buquiterine, buramate, burodiline, buspirone, busulfan, butabarbital, butacaine, butacetin, butaclamol, butadiazamide, butafosfan, butalamine, butalbital, butamben, butamirate, butamisole, butamoxane, butanediol cyclic sulfite, butanilicaine, butanixin, butanserin, butantrone, butaperazine, butaprost, butaverine, butedronate, buterizine, butetamate, butethamine, buthiazide, butibufen, butidrine, butikacin, butilfenin, butinazocine, butinoline, butirosin, butixirate, butobendine, butoconazole, butocrolol, butoctamide, butofilolol, butonate, butopamine, butopiprine, butoprozine, butopyrammonium iodide, butorphanol, butoxamine, butoxylate, butriptyline, butropium bromide, butylated hydroxyanisole, butylated hydroxytoluene, butylparaben, butynamine, buzepide metiodide, cabastine, cabergoline, cadralazine, cafaminol, cafedrine, caffeine, calcifediol, calcitrol, calcium citrate, calcium dobesilate, calcium glubionate, calcium gluceptate, calcium gluconate, calcium glycerophosphate, calcium hypophosphite, calcium lactate, calcium lactobionate, calcium levulinate, calcium mandelate, calcium pantothenate, calcium phosphate dibasic, calcium phophate tribasic, calcium saccharate, calcium stearate, calusterone, camazepam, cambendazole, camiverine, camostast, camphotamide, camptothecin, camylofin, canbisol, cannabinol, canrenoic acid, canrenone, cantharidine, capobenic acid, capreomycin, caproxamine, capsaicine, captamine, captodiame, captopril, capuride, caracemide, caramiphen, carazolol, carbachol, carbadox, carbaldrate, carbamazepine, carbamide peroxide, carbantel lauryl sulfate, carbaril, carbarsone, carbaspirin calcium, carbazeran, carbazochrome, carbazachrome salicylate, carbazachrome sulfonate, carbazocine, carbeniciltin, carbenicillin indanyl, carbencillin phenyl, carbenoxolone, carbenzide, carbestrol, carbetapentane, carbidopa, carbimazole, carbinoxamine, carbiphene, carbocloral, carbocysteine, carbofenotion, carbol-fuschin, carbomycin, carboplatin, carboprost, carboprost methyl, carboquone, carbromal, carbubarb, carburazepam, carbutamide, carbuterol, carcainium chloride, carebastine, carfentanil, carfimate, carisoprodol, carmantadine, carmetizide, carmofur, carmustine, carnidazole, carnitine, carocainide, caroverine, caroxazone, carperidine, caperone, carphenazine, carpindolol, carpiramine, carprofen, carpronium chloride, carsalam, cartazolate, carteolol, carubicin, carumonam, carvedilol, carzenide, carzolamide, cathine, cathinone, cefaclor, cefadroxil, cefaloniumi, cefaloram, cefamandole, cefamandole naftate, cefaparole, cefatrizine, cefazaflur, cefazedone, cefazolin, cefbuperazone, cefcanel, cefcanel daloxate, cefedrolor, cefempidone, cefepime, cefetamet, cefetrizole, cefvitril, cefixime, cefmenoxime, cefmepidium chloride, cefmetazole, cefminox, cefodizime, cefonizid, cefoperazone, ceforanide, cefotaxime, cefotetan, cefotiam, cefoxazole, cefoxitin, cefpimizole, cefpiramide, cefpirome, cefpodoxime, cefpodoxime proxetil, cefquinome, cefrotil, cefroxadine, cefsulodin, cefsumide, ceftazidime, cefteram, ceftezole, ceftiofur, ceftiolene, ceftioxide, ceftizoxime, ceftriaxone, cefuracetime, cefuroxime, cefuraxime axetil, cefurzonam, celiprolol, cephacetrile, cephalexin, cephaloglycin, cephaloridine, cephalothin, cephapirin, cephradine, cetaben, cetamolol, cethexonium chloride, cetiedil, cetirizine, cetocycline, cetohexazine, cetophenicol, cetotiamine, cetoxime, cetraxate, chaulmosulfone, chendiol, chiniofon, chlophedianol, chloracyzine, chloral betaine, chloral hydrate, chloralose, chlorambucil, chloramine, chloramphenicol, chloramphenicol palmitate, chloramphenicol succinate, chlorazanil, chlorbenzoxamine, chlorbetamide, chlorcyclizine, chlordantoin, chlordiazepoxide, chlordimorine, chlorhexadol, chlorhexidine, chlorhexidine phosphanilate, chlorindanol, chlorisondamine chloride, chlormadinone acetate, chlormerodrin, chlormezanone, chlormidazole, chloronaphazine, chloroazodin, chlorobutanol, chlorocresol, chlorodihydroxyandrostenone, chloroethyl mesylate, 5-chloro-3′-fluoro-2′3-dideoxyuridine, chloroguanide, chlorophenothane, chloroprednisone acetate, chloroprocaine, chloropyramine, chloroquine, chloroserpidine, chlorothen, chlorothiazide, chlorotriansene, chloroxine, chloroxylenol, chlorozotocin, chlorphenesin, chlorphenesin carbamate, chlorpheniramine, chlorphenoctium amsonate, chlorphenoxamine, chlorphentermine, chlorproethazine, chlorproguanil, chlorpromazine, chlorpropamide, chlorprothixene, chlorquinaldol, chlortetracycline, chlorthalidone, chlorthenoxazine, chlorzoaxazone, chloecalciferol, cholic acid, choline chloride, choline glycerophosphate, chromocarb, chromonar, ciadox, ciamexon, cianergoline, cianidol, cianopramine, ciapilome, cicaprost, cicarperone, ciclactate, ciclafrine, ciclazindol, cicletanine, ciclomenol, ciclonicate, ciclonium bromide, ciclopirox, ciclopramine, cicloprofen, cicloprolol, ciclosidomine, ciclotizolam, ciclotropium bromide, cicloxilic acid, cicloxolone, cicortonide, cicrotic acid, cidoxepin, cifenline, cifostodine, ciglitazone, ciheptolane, ciladopa, cilastatine, cilazapril, cilazaprilat, cilobamine, cilofungin, cilostamide, cilostazol, ciltoprazine, cimaterol, cimemoxin, cimepanol, cimetidine, cimetropium bromide, cimoxatone, cinchonine, cinchophen, cinecromen, cinepaxadil, cinepazet, cinepazic acid, cinepazide, cinfenine, cinfenoac, cinflumide, cingestol, cinitapride, cinmetacin, cinnamaverine, cinnamedrine, cinnarizine, cinnarizine clofibrate, cinnofuradione, cincotramide, cinodine, cinolazepam, cinoquidox, cinoaxin, cinoxate, cinoxolone, cinooxopazide, cinperene, cinprazole, cinpropazide, cinromide, cintazone, cintriamide, cinperone, ciprafamide, ciprafazone, ciprefadol, ciprocinonide, ciprofibrate, ciprofloxacin, cipropride, ciproquazone, ciprostene, ciramadol, cirazoline, cisapride, cisconazole, cismadinone, cisplatin, cistinexine, citalopram, citatepine, citenamide, citenazone, citicoline, citiolone, clamidoxic acid, clamoxyquin, clanfenur, clanobutin, clantifen, clarithromycin, clavulanic acid, clazolam, clazolimine, clazuril, clebopride, clefamide, clemastine, clemeprol, clemizole, clenbuterol, clenpirin, cletoquine, clibucaine, clidafidine, clidanac, clidinum bromide, climazolam, climbazole, climiqualine, clindamycin, clindamycin palmitate, clindamycin phosphate, clinofibrate, clinolamide, cliquinol, clioxanide, clipoxamine, cliprofen, clobazam, clobenoside, clobenzepam, clobenzorex, clobenztropine, clobetasol propionate, clobetasone butyrate, clobutinol, clobuzarit, clocanfamide, clocapramine, clociguanil, clocinizine, clocortolone acetate, clocortolone pivalate, clocoumarol, clodacaine, clodanolene, clodazon, clodoxopone, clodronic acid, clofazimine, clofenamic acid, clofenamide, clofenciclan, clofenetamine, clofenoxyde, clofenvinfos, clofeverine, clofexamide, clofezone, clofibrate, clofibric acid, clofibride, clofilium phosphate, cloflucarban, clofoctol, cloforex, clofurac, clogestone acetate, cloguanamil, clomacran, clomegestone acetate, clometacin, clometherone, clomethiazole, clometocillin, clomifenoxide, clominorex, clomiphene, clomipramine, clomocycline, clomoxir, clonazepam, clonazoline, clonidine, clonitazene, clonitrate, clonixeril, clonixin, clopamide, clopenthixol, cloperastine, cloperidone, clopidogrel, clopidol, clopimozide, clopipazan, clopirac, cloponone, cloprednol, cloprostenol, cloprothiazole, cloquinate, cloquinozine, cloracetadol, cloranolol, clorazepate, clorethate, clorexolone, clorgiline, cloricromen, cloridarol, clorindanic acid, clorindione, clormecaine, cloroperone, clorophene, cloroqualone, clorotepine, clorprenaline, clorsulon, clortermine, closantel, closiramine, clostebol, clothiapine, clothixamide, clotiazepam, cloticasone propionate, clotioxone, clotrimazole, clovoxamine, cloxacepride, cloxacillin, cloxacillin benzathine, cloxazolam, cloxestradiol, cloximate, cloxotestosterone, cloxypendyl, cloxyquin, clozapine, cobamide, cocaine, cocarboxylase, codeine, codoxime, cofisatin, cogazocine, colchicine, colestolone, colfenamate, colforasin, colterol, conessine, conorphone, copper gluconate, cormethasone acetate, corticosterone, cortisone acetate, cortisuzol, cortivazol, cortodoxone, cotarnine chloride, cotinine, cotriptyline, coumaphos, coumazoline, coumermycin, coumetarol, creatinolfosfate, crisnatol, croconazole, cromakalim, cromitrile, cromolyn, cropropamide, crospovidone, crotamiton, crotetamide, crotoniazide, crufomate, cuprimyxin, cuproxoline, cyacetacide, cyamemazine, cyanocobalamine, cyclacillin, cyclandelate, cyclarbamate, cyclazocine, cyclazodone, cyclexanone, cyclindole, cycliramine, cyclizine, cyclobarbital, cyclobendazole, cyclobenzaprine, cyclobutoic acid, cyclobutyrol, cyclofenil, cycloguanil, cloheximide, cycloleucine, cyclomenol, cyclomethicone, cyclomethycaine, cyclopentamine, cyclopenthiazide, cyclopentolate, cyclopenazine, cyclophosphamide, cyclopregnol, cyclopyrronium bromide, cycloserine, cyclosporine, cyclothiazide, cyclovalone, cycotiamine, cycrimine, cyheptamid, cyheptropine, cynarine, cypenamine, cypothrin, cyprazepam, cyprenophine, cyprodenate, cyproheptadine, cyprolidol, cyproquinate, cyproterone acetate, cyproximide, cystine, cytarabine, dacarbazine, dacemazine, dacisteine, dacinomycin, dacuronium bromide, dagapamil, dalbraminol, daledalin, daltroban, dametralast, damotepine, danazol, danitracen, danosteine, danthron, dantrolene, dapiprazole, dapsone, daptomycin, darenzepine, darodipine, datelliptium chloride, dunorubicin, dazadrol, dazepinil, dazidamine, dazmegrel, dazolicine, dazopride, dazoquinast, dacoxiben, deanol aceglumate, deanol acetaminobenzoate, deazauridine, deboxamet, debrisoquin, decamethonium bromide, decimemide, decitropine, declaben, declenperone, decloxizine, decominol, decoquinate, deditonium bromide, deferoxamine, deflazacort, defosfamide, dehydroacetic acid, dehydroemetine, dehydro-7-methyltestosterone, delanterone, delapril, delergotrile, delfantrine, delmadinone acetate, delmetacin, delmopinol, delorazepam, deloxone, delprostenate, dembrexine, demecarium bromide, demeclocycline, demecolcine, demecycline, demegestone, demelverine, demexiptiline, democonazole, demoxepam, denaverine, denbufylline, denipride, denopamine, denpidazone, denzimol, deoxyspergualin, depramine, deprodone, deprostil, deptropine, derpanicate, desacetylcolchicine tartrate, desaspidin, desiclovir, descinolone acetonide, deserpidine, desipramine, deslanoside, desmethylcolchicine, desmethylmisonidazole, desmethylmoramide, desocriptine, desogestrel, desomorphine, desonide, desoximetasone, desoxycorticosterone acetate, desoxycorticosterone pivalate, desoxypyridoxine, detajmium bitartrate, detanosal, deterenol, detomidine, detorubicin, detrothronine, devapamil, dexamethasone, dexamethasone, acefurate, dexamethasone acetate, dexamethasone dipropionate, dexamethasone phosphate, dexamisole, dexbrompheniramine, dexchlorpheniramine, dexclamol, dexetimide, dexetozolin, dexfenfluramine, deximafen, dexindoprofen, dexivacaine, dexlofexidine, dexmedetomidine, dexoxadrol, dexpanthenol, dexpropranolol, dexproxibutene, dexecoverine, dextilidine, dextroamphetamine, dextrofemine, dextromethorphan, dextromoramide, dextrorphan, dextrothyroxine, dezaguanine, dezocine, diacerein, diacetamate, diacetolol, diacetylmorphine, diamfenetide, diaminomethylphenazinium chloride, diamocaine, diampromide, diamthazole, dianhydrogalactitol, diapamide, diarbarone, diathymosulfone, diatrizoic acid, diaveridine, diazepam, diaziquone, diazoacetylglycine hydrazide, diazouracil, diazoxide, dibekacin, dibemethine, dibenamine, dibenzepin, dibrompropamidine, dibromsalan, dibrospidium chloride, dibucaine, dibuprol, dibupyrone, dibusadol, dicarbine, dicarfen, dichlorallyl lawsone, dichlorisone acetate, dichlormezanone, dichlorofluormethane, dichloromethotrexate, dichlorophen, dichlorophenarsine, dichlorotetrafluoroethane, dichloroxylenol, dichlorphenamide, dichlorvos, diciferron, dicirenone, diclazuril, diclofenac, diclofensine, diclofurime, diclometide, diclonixin, dicloxacillin, dicobalt edetate, dicolinium iodide, dicresulene, dicumarol, dicyclomine, didemnin, dideoxycytidine, didrovaltrate, dieldrin, dienestrol, dienogest, diethadione, diethazine, diethylpropion, diethylstilbestrol, diethylstilbestrol diphosphate, diethylstilbestrol dipropionate, diethylthiambutene, diethyltoluamide, dietifen, difebarbamate, difemerine, difemetorex, difenamizole, difencloxazine, difenoximide, difenoxin, difetarsone, difeterol, diflorasone diacetate, difloxacin, difluanine, diflucortolone, diflurcortolone pivalate, diflumidone, diflunisal, difluprednate, diftalone, digalloyl trioleate, digitoxin, digoxin, dihexyverine, dihydralazine, dihydroazacytidine, dihydroergotamine, dihydrolenperone, dihydrostreptomycin, dihydrotachysterol, dihydroxyfluoroprogestrone, diisopromine, diisopropanolamine, dilazep, dilevalol, dilmefone, diloxanide, diltiazem, dimabefylline, dimecamine, dimecolonium iodide, dimecrotic acid, dimefadane, dimefline, dimelazine, dimemorfan, dimenhydrinate, dimenoxadol, dimeheptanol, dimepranol, dimepregnen, dimeprozan, dimercaprol, dimesna, dimesone, dimetacrine, dimetamfetamine, dimethadione, dimethaminostyrylquinoline, dimethazan, dimethindene, dimethiodal, dimethisoquin, dimethisterone, dimetholizine, dimethoxanate, dimethylhydroxytestosterone, dimethylnorandrostadienone, dimethylnortestosterone, dimethylstilbestrol, dimethyl, dimethylthiambutene, dimethyltubocurarinium chloride, dimetipirium bromide, dimetofrine, dimetridazole, diminazene, dimoxamine, dimoxaprost, dimoxyline, dimpylate, dinaline, dinazafoqe, diniprofylline, dinitolmide, dinoprost, dinoprostone, dinsed, diosmin, dioxadilol, dioxadrol, dioxamate, dioxaphetyl butyrate, dioxethedrin, dioxifedrine, dioxybenzone, dipenine bromide, diperodon, diphemanil methylsulfate, diphenadione, diphenan, diphenhydramine, diphendiol, diphenoxylate, diphenylpraline, diphoxazide, dipipanone, dipipoverine, dipiverin, diprafenone, diprenorphine, diprobutine, diprofene, diprogulic acid, diproleandomycin, diproqualone, diproteverine, diprotriozate, diproxadol, dipyridamole, dipyrithione, dipyrocetyl, dipyrone, dirithromycin, disobutamide, disofenin, disogluside, disopyramide, disoxaril, distigmine bromide, disulergine, disulfamide, disulfiram, disuprazole, ditazole, ditercalinium chloride, dithiazanine iodide, ditiocarb, ditiomustine, ditolamide, ditophal, divabuterol, dixanthogen, dizatrifone, dizocilpine, dobupride, dobutamine, docarpamine, doconazole, docusate, doliracetam, domazoline, domiodol, domiphen bromide, domipizone, domoprednate, domoxin, domperidone, don, donetidine, dopamantine, dopamine, dopexamine, dopropidil, doqualast, dorastine, doreptide, dosergoside, dotarizine, dotefonium bromide, dothiepin, doxacurium chloride, doxaminol, doxapram, doxaprost, doxazosin, doxefazepam, doxenitoin, doxepin, doxibetasol, doxifluridine, doxofylline, doxorubicin, doxpicomine, doxycycline, doxylamine, dramedilol, draquinolol, deazidox, dribendazole, drindene, drobuline, drocinonide, droclidinium bromide, drocode, drofenine, droloxifene, drometrizole, dromostanolone, dromostanolone propionate, dronabinol, dropempine, droperidol, droprenilamine, dropropizine, drotaverine, drotebanol, droxacin, droxicainide, droxicam, droxidopa, droxypropine, dulofibrate, dulozafone, duometacin, duoperone, dupracetam, durapatite, dyclonine, dydrogesterone, dymanthine, dyphylline, ebastine, ebrotidine, ebselen, ecastolol, echinomycin, echothiophate iodide, ecipramidil, eclanamine, eclazolast, econazole, ectylurea, edelfosine, edetic acid, edetol, edifolone, edogestrone, edoxudine, edrophonicum chloride, efaroxan, efetozole, eflornithine, efloxate, efrotomycin, elantrine, elanzepine, elderfield's pyrimidine mustard, elfazepam, ellagic acid, elliptinium acetate, elmustine, elnadipine, eltenac, eltoprazine, elucaine, elziverine, embramine, embutramide, emepronium bromide, emetine, emiglitate, emilium tosylate, emopanil, emorfazone, emylcamate, enalapril, enalaprilat, enbucrilate, encainide, enciprazine, enclomiphene, encyprate, endomide, endralazine, endrysone, enefexine, enestebol, enfenamic acid, enflurane, eniclobrate, enilconazole, enilospirone, enisoprost, enocitabine, enolicam, enoxacin, enoxamast, enoximone, enoxolone, eniprazole, eniproline, enprazepine, enprofylline, enpromate, enprostil, enrofloxacin, entsufon sodium, enviomycin, enviradene, epalretat, epanolol, eperisone, ephedrine, epicainide, epicillin, epicriptine, epiestriol, epimestrol, epinastine, epinephrine, epinephryl borate, epipropidine, epirizole, epiroprim, epirubicin, epithiazide, epitiostanol, epoprostenol, epostane, eprazinone, eprovafen, eproxindine, eprozinol, epsiprantel, eptaloprost, eptazocine, equilin, erdosteine, ergocalciferol, ergoloid mesylates, ergonovine, ergosterol, ergotamine, ericolol, erizepine, erocainide, erythrityl tetranitrate, erythromycin, erythromycin acistrate, erythromycin ethylsuccinate, erythromycin propionate, erythrosine, esaprazole, esculamine, eseridine, esflurbiprofen, esmolol, esorubicin, esproquin, estazolam, estradiol, estradiol benzoate, estradiol cypionate, estradiol dipropionate, estradiol enanthate, estradiol undecylate, estradiol valerate, estramustine, estramustine phosphate, estrapronicate, estrazinol, estriol, estrofurate, estrone, estrone hydrogen sulfate, estropipate, esuprone, etabenzarone, etacepride, etafedrine, etafenone, etamestrol, etamiline, etamiphyllin, etamocycline, etanidazole, etanterol, etaqualone, etasuline, etazepine, etazolate, etebenecid, eterobarb, etersalate, ethacridine, ethacrynic acid, ethambutol, ethamivan, ethamsylate, ethanolamine oleate, ethaverine, ethchlorvynol, ethenzamide, ethazide, ethidium chloride, ethinamate, ethinyl estradiol, ethiofos, ethionamide, ethsterone, ethoheptazine, ethomoxane, ethonam, ethopropazine, ethosuximide, ethotoin, ethoxazene, ethoxazorutoside, ethoxzolamide, ethyybenztropine, ethyl biscoumacetate, ethyl carfluzepate, ethyl cartrizoate, ethyl dibunate, ethyl dirazepate, ethylenediamine, ethylestrenol, ethylhydrocupreine, ethyl loflazepate, ethylmethylthiambutene, ethylmorphine, 9-ethyl-6-mercaptopurine, ethyl nitrite, ethylnorepinephrine, ethylparaben, ethylphenacemide, ethylstibamine, ethynerone, ethynodiol diacetate, ethypicone, etibendazole, eticlopride, eticyclidine, etidocaine, etidronic acid, etifelmine, etifenin, etifoxine, etilamfetamine, etilefrine, etilefrin pivalate, etintidine, etiochlanolone, etipirium iodide, etiproston, etiracetam, etiroxate, etisazole, etisomicin, etisulergine, etizolam, etocarlide, etocrylene, etodolac, etodroxzine, etofamide, etofenamate, etofenprox, etofibrate, etoformin, etofuradine, etofylline, etoglucid, etolorex, etolotifen, etoloxamine, etomidate, etomidoline, etomoxir, etonitazene, etoperidone, etoposide, etoprindole, etoprine, etorphine, etosalamide, etoxadrol, etoxeridine, etozolin, etrabamine, etretinate, etryptamine, etymemazine, eucalyptol, eucatropine, eugenol, euprocin, evandamine, Evans blue, exalamide, exametazine, exaprolol, exepanol, exifone, exiproben, falintolol, falipamil, famiraprinium chloride, famotidine, famotine, famiprofazone, fanetizole, fantridone, fazadinium bromide, fazaribine, febantel, febarbamate, februpol, febuverine, feclemine, feclobuzone, fedrilate, felbamate, felbinac, felipyrine, felodipine, femoxetine, fenabutene, fenacetinol, fenaclon, fenadiazole, fenaptic acid, fenalamide, fenalcomine, fenamifuril, penamole, fenaperone, fenbendazole, fenbencillin, fenbufen, fenbutrazate, fencamfamine, fencibutirol, fenclexonium metilsulfate, fenclofen4c, fenclonine, fenclorac, fenlozic acid, fendiline, fendosal, feneritrol, fenestrel, fenethazine, fenethylline, fenetradil, fenflumizole, fenfluramine, fenfluthrin, fengabine, fenharmane, fenimide, feniodium chloride, fenipentol, fenirofibrate, fenisorex, fenmetozole, fenmetramide, fenobam, fenocinol, fenoctimine, fenofibrate, fenoldopam, fenoprofen, fenoterol, fenoverine, fenoxazoline, fenoxedil, fenozolone, fenpentadiol, fenperate, fenipalone, fenipramide, feniprane, fenpiverinium bromide, fenprinast, fenproporex, fenprostalene, fenquizone, fenretinide, fenspiride, fentanyl, fentiazac, fenticlor, fenticonazole, fentonium bromide, fenyripol, fepentolic acid, fepitrizol, fepradinol, feprazone, fepromide, feprosidnine, ferriclate calcium, ferrotrenine, ferrous fumarate, ferrous gluconate, fetoxylate, fexicaine, fexinidazole, fezatione, fezolamine, fiacitabine, fibracillin, filenadol, filipin, fifexide, flamenol, flavamine, flavodic acid, flavodil, flavoneactic acid, flavoxate, flazalone, flecainide, flerobuterol, fleroxacin, flesinoxan, flestolol, fletazepam, floctafenine, flomoxef, flopropione, florantyrone, flordipine, floredil, florfenicol, florifenine, flosequinan, flotrenizine, floverine, floxacillin, floxacrine, floxuridine, fluacizine, flualamide, fluanisone, fluazacort, flubanilate, flubendazole, flubepride, flucabril, flucetorex, flucindole, fluciprazine, flucloronide, fluconazole, flucrylate, flucytosine, fludalanine, fludarabine phosphate, fludazonium chloride, fludiazepam, fludorex, fludoxopone, fludrocortisone acetate, flufenamic acid, flufenisal, flufosal, flufylline, fluindarol, fluindione, flumazenil, flumecinol, flumedroxone-17-acetate, flumequine, flumeridone, flumethasone, flumethasone pivalate, flumethiazide, flumetramide, flumexadol, flumezapine, fluminorex, flumizole, flumoxonide, flunamine, flunarizine, flunidazole, flunisolide, flunisolide acetate, flunitrazepan, flunixin, flunoprost, flunoxaprofen, fluocinolone acetonide, fluocinonide, flourcortin butyrate, fluocortolone, fluocortolone caproate, fluorescein, fluoresone, fluoroadenosine, 3-fluoroandrostanol, fluorodopane, fluorohydroxyandrosterone, fluorometholone, fluorometholone acetate, fluorosalan, 6-fluorotestosterone propionate, fluorouracil, 9-fluoroxotestenololactone, 9-fluoroxotestololacetone, fluotracen, fluoxetine, fluoxymesterone, fluparoxan, flupentixol, fluperamide, fluperlapine, fluperolone acetate, fluphenazine, fluphenazine enanthate, flupimazine, flupirtine, flupranone, fluprazine, fluprednidene, fluprednisolone, fluprednisolone valerate, fluprofen, fluprofylline, fluproquazone, fluprostenol, fluquazone, fluradoline, flurandrenoline, flurantel, flurazepam, flurbiprofen, fluretofen, flurithromycin, flurocitabine, flurofamide, flurogestone acetate, flurothyl, fluroxene, flusoxolol, fluspiperone, fluspirilene, flutamide, flutazolam, flutemazepam, flutiazin, fluticasone propionate, flutizenol, flutonidine, flutoprazepam, flutroline, flutropium bromide, fluvoxamine, fluzinamide, fluzoperine, folescutol, folic acid, fomidacillin, fominoben, fomocaine, fonazine, fopirtoline, forfenimex, formebolone, formetorex, formintrazole, formocortal, formoterol, fosarilate, fosazepam, foscarnet, foscolic acid, fosenazide, fosfocreatine, fosfomycin, fosfonet, fosfosal, fosinapril, fosmenic acid, fosmidomycin, forpirate, fostedil, fostriecin, fotemustine, fotreamine, frabuprofen, frentizole, fronepidil, froxiprost, ftaxilide, ftivazide, ftorafur, ftormetazine, ftorpropazine, fubrogonium iodide, fuchsin, fumagillin, fumoxcillin, fuprazole, furacrinic acid, furafylline, furalazine, furaltadone, furaprofen, furazabol, furazolidone, furazolium chloride, furbucillin, furcloprofen, furegrelate, furethidine, furfenorex, furidarone, furmethoxadone, furobufen, furodazole, furofenac, furomazine, furosemide, furostilbestrol, fursalan, fursuItiamine, furterene, furtrethonium iodide, fusidic acid, fuzlocillin, gabapentin, gabexate, gaboxadol, galantamine, gallamine triethodide, gallopamil, galosemide, galtifenin, gampexine, gamolenic acid, ganciclovir, ganglefene, gapicomine, gapromidine, gefarnate, gemazocine, gemcadiol, gemeprost, gemfibrozil, gentamicin, gentian violet, gepefrine, gepirone, geroquinol, gestaclone, gestadienol, gestodene, gestonorone caproate, gestrinone, giparmen, gitaloxin, gitoformate, glafenine, glaziovine, gliamilide, glibornuride, glibutimine, glicaramide, glicetanile, geroquinol, gestaclone, gestadienol, gestodene, gestonorone caproate, gestrinone, giparmen, gitaloxin, gitoformate, glafenine, glaziovine, gliamilide, glibornuride, glibutimine, glicaramide, glicetanile, gliclazide, glicondamide, glidazamide, gliflumide, glimepiride, glipentide, glipizide, gliquidone, glisamuride, glisindamide, glisolamide, glisoxepide, gloxazone, gloximonam, glucametacin, glucosamine, glucosulfamide, glucosulfone, glucurolactone, glucuronamide, glunicate, glutamic acid, glutaral, glutarimide, glutaurine, glutethimide, glyburide, glybuthiazol, glybuzole, glyceryl monostearate, glycidyl methacrylate, glycine, glyclopyramide, glybiarsol, glycopyrrolate, glycyclamide, glyhexamide, glymidine, glyoctamide, glypinamide, glyprothiazol, glysobuzole, gold thiomalate, gold sodium thiosulfate, granisetron, griseofulvin, guabenxan, guacetisal, guafecainol, guaiactamine, guaiapate, guaietolin, guaifenesin, guaimesal, guaisteine, guaithylline, guamecycline, guanabenz, guanacline, guanadrel, guanazodine, guanazole, guanclofine, guancydine, guanethidine, guanfacine, guanisoquin, guanoclor, guanoctine, guanoxabenz, guanoxan, guanoxyfen, hadacidin, halazepam, halazone, halcinonide, halethazole, halocortolone, halofantrine, halofenate, halofuginone, halometasone, halonamine, halopemide, halopenium chloride, haloperidol, haloperidol decanoate, haloperidone acetate, haloprogesterone, haloprogin, halothane, haloxazolam, haloxon, halquinols, hedaquinium chloride, hepronicate, heptabarbital, heptaminol, heptaverine, heptolamide, hepzidine, hetacillin, hetaflur, heteronium bromide, hexachlorophene, hexacyclonate, hexacyprone, hexadiline, hexadimethrine bromide, hexafluorenium bromide, hexamethonium bromide, hexamidine, hexapradol, hexaprofen, hexapropymate, hexasonium iodide, hexacarbacholine bromide, hexedine, hexestrol, hexetidine, hexobarbital, hexobendine, hexocyclium methylsulfate, hexoprenaline, hexopyrronium bromide, hexylcaine, hexylene glycol, hexylresorcinol, histamine, histapyrrodine, homarylamine, homatropine, homatropine methylbromide, homidium bromide, homochlorcyclizine, homofenazine, homoharringtonine, homopipramol, homosalate, homotestosterone propionate, homprenorphine, hopantenic acid, hoquizil, hycanthone, hydracarbazine, hydralazine, hydrargaphen, hydrobentizide, hydrochlorthiazide, hydrocodone, hydrocortamate, hydrocortisone, hydrocortisone aceponate, hydrocortisone acetate, hydrocortisone butyrate, hydrocortisone cypionate, hydrocortisone-phosphate, hydrocortisone succinate, hydrocortisone valerate, hydroflumethiazide, hydromadinone, hydromorphinol, hydromorphone, hydroquinone, hydroxindasate, hydroxindasol, hydroxyoxocobalamin, hydroxy amphetamine, hydroxychloroquine, hydroxydimethandrostadienone, hydroxydione succinate, hydroxymethylandrostanone, 10-hydroxynorehisterone, hydroxypethidine, hydroxyphenamate, hydroxyprocaine, hydroxyprogeserone, hydroxyprogesterone caproate, hydroxypyridine tartrate, hydroxystilbamidine, 7-hydroxytestololacetone, hydroxytestosterone propionate, hydroxytetracaine, hydroxytoluic acid, hydroxyurea, hydroxyzine, hymecromone, hyoscyamine, hypericin, ibacitabine, ibafloxacin, ibazocine, ibopamine, ibrotamide, ibudilast, ibufenac, ibuprofen, ibuprofen piconol, ibuproxam, ibuterol, ibuverine, icazepam, icosipiramide, icotidine, idarubicin, idaverine, idazoxan, idebenone, idenast, idoxuridine, idralfidine, idrocilamide, idropranolol, ifenprodil, ifosfamide, ifoxetine, ilmofosine, iloprost, imafen, imanixil, imazodan, imcarbofos, imexon, imiclopazine, imidazole salicylate, imidazopyrazole, imidecyl iodine, imidocarb, imidoline, imidurea, imiloxan, iminophendimide, imipenem, imipramine, imipraminoxide, imirestat, imolamine, imoxiterol, impacarzine, impromidine, improsulfan, imuracetam, inaperisone, indacrinone, indalpine, indanazoline, indanidine, indanorex, indapamide, indatraline, indacainide, indeloxazine, indenolol, indicine-N-oxide, indigotindisulfonic acid, indobufen, indocate, indocyanine green, indolapril, indolidan, indomethacin, indopanolol, indopine, indoprofen, indoramin, indorenate, indoxole, indriline, inicarone, inocoterone, inosine, inosine dialdehyde, inositol niacinate, inproquone, intrazole, intriptyline, iobenzamic acid, iobutic acid, iocarmic acid, iocetamic acid, iodamide, iodecimol, iodetryl, iodipamide, iodixanol, iodoalphionic acid, iodol, iodophthalein, iodoquinol, iodothiouracil, iodoxamic acid, ioglicic acid, ioglucol, ioglucomide, ioglunide, ioglycamic acid, iogulamide, iohexol, iodlidonic acid, iolixanic acid, iomeglamic acid, iomeprol, iomorinic acid, iopamidol, iopanoic acid, iopentol, iophendylate, iophenoxic acid, ioprocemic acid, iopromide, iopronic acid, iopydol, iopydone, iosarcol, iosefamic acid, ioseric acid, iosimide, iosulamide, iosumetic acid, iotasul, iotetric acid, iothalamic acid, iotranic acid, iotrizoic acid, iotrolan, iotroxic acid, ioversol, ioxabrolic acid, ioxaglic acid, ioxitalamic acid, ioxotrizoic acid, iozomic acid, ipexidine, ipodic acid, ipragratine, ipramidil, ipratropium bromide, iprazochrome, ipriflavone, iprindole, iprocinodine, iproclozide, iprocrolol, iprofenin, iproheptine, iproniazid, iproidazole, iproplatin, iprotiazem, iproxamine, iprozilamine, ipsalazide, ipsapirone, iquindamine, irindalone, irloxacin, irolapride, irsogladine, isamfazone, isamoltan, isamoxole, isaxonine, isbogrel, isepamicin, isoaminile, isobromindione, isobucaine, isobutamben, isocarboxazid, isoconazole, isocromil, isoetharine, isofezolac, isoflupredone acetate, isoflurane, isoflurophate, isoleucine, isomazole, isomerol, isometamidium, isomethadone, isomethept ne, isomylamine, isoniazid, isonixin, isoprazone, isoprednidene, isoprofen, isoprofamide iodide, isopropicillin, isopropyl myristate, isopropyl palmitate, isoproterenol, isosorbide, isosorbide dinitrate, isosorbide mononitrate, isospalglumic acid, isosulfan blue, isosulpride, isothipendyl, isotic, isotiquimide, isotretinoin, isoxaprolol, isoxepac, isoxicam, isoxsuprine, isradipine, itanoxone, itazigrel, itraconazole, itrocainide, ivermectin bib, ivoqualine, josamycin, kainic acid, kalafungin, kanamycin, kebuzone, keracyanin, ketamine, ketanserin, ketazocine, ketazolam, kethoxal, ketipramine, ketobemidone, ketocaine, ketocainol, ketoconazole, ketoprofen, ketorfanol, ketorolac, ketotifen, ketotrexate, khellin, khelloside, kitasamycin, labetalol, lacidipine, lactalfate, lactose, lactulose, lamotrigine, lamtidine, lanatoside, lapachol, lapinone, lapyrium chloride, la, salocid, laudexium methyl sulfate, lauralkonium chloride, laureth, laurixamine, laurocapram, lauroguadine, laurolinium acetate, lauryl isoquinolinium, lefetamine, leflunomide, leiopyrrole, lemidosul, lenampicillin, leniquinsin, lenperone, leptacline, lergotrile, letimide, letosteine, leucine, leucinocaine, leucocianidol, leucovorin, levacecarnine, levallorphan, levamfetamine, levamisole, levdropropizine, levisoprenaline, levlofexidine, levobunolol, levocabastine, levocarnitine, levodopa, levofacetoperane, levofenfluramine, levofuraltadone, levoglutamide, levomenol, levomethadone, levomethadyl acetate, levomethorphan, levometiomeprazine, levomopranol, levomoramide, levonantradol, levonordeprin, levonorgestrel, levophenacyl morphan, levopropoxyphene, levopropylcillin, levopropylhexedrine, levoprotiline, levorin, levorphanol, levothyroxine, levoxadrol, lexofenac, libecillide, libenzapril, lidamidine, lidocaine, lidofenin, lidoflazine, lifibrate, lilopristone, limaprost, lincomycin, lindane, linsidomine, liothyronine, liroldine, lisinopril, lisuride, lithium carbonate, lithium citrate, litracen, lividomycin, lixazinone, lobeline, lobendazole, lobenzarit, lobuprofen, locicortone, lodaxaprine, lodacezarlodinixil, lodiperone, lodoxamide, lodoxamide ethyl, lofemizole, lofendazam, lofentanil, lofepramine, lofexidine, loflucarban, lombazole, lomefloxacin, lometraline, lomevactone, lomifylline, lomofungin, lomustine, lonapalene, lonaprofen, lonazolac, lonidamine, loperamide, loperamide oxide, lopirazepam, loprazolam, loprodiol, lorajmine, lorapride, loratadine, lorazepam, lorbamate, lorcainide, lorcinadol, lorglumide, lormetazepam, lortalamine, lorzafone, losindole, losulazine, lotifazole, lotrifen, lotucaine, lovastatin, loxanast, loxapine, loxiglumide, loxoprofen, loxtidine, lozilurea, lucanthone, lucartamide, lucimycin, lufuradom, lupitidine, luprostiol, luxabendazole, lyapolate sodium, lycetamine, lydimycin, lymecycline, lynestrenol, lysergide, lysine, mabuterol, maduramicin, mafenide, mafoprazine, mafosfamide, magnesium citrate, magnesium, gluconate, magnesium salicylate, malathion, malethamer, malic acid, malotilate, manidipine, manganese gluconate, mannitol, mannitol hexanitrate, mannomustine, mannosulfan, manozodil, maprotiline, maridomycin, mariptiline, maroxepin, maytansine, mazaticol, mazindol, mazipredone, mebanazine, mebendazole, mebenoside, mebeverine, mebezonium iodide, mebhydrolin, mebiquine, mebolazine, mebrofenin, mebutamate, mebutizide, mecamylamine, mecarbinate, mecetronium ethylsulfate, mechlorethamine, meciadanol, mecinarone, meclizine, meclocycline, meclocycline sulfosalicylate, meclofenamic acid, meclofenoxate, meclonazepam, mecloqualone, mecloralurea, meclorisone dibutyrate, mecloxamine, mecobalamin, mecrylate, mecysteine, medazepam, medazomide, medetomidine, medibazine, medifoxamine, medorinone, medorubicin, medrogestone, medronic acid, medroxalol, medroxyprogestrone, medroxyprog strone acetate, medrylamine, medrysone, mefeclorazine, mefenamic acid, mefenidil, mefenidramium metilsulfate, mefenorex, mefeserpine, mefexamide, mefloquine, mefruside, megalomicin, megestrol acetate, meglitinide, megucycline, meglumine, meglutol, meladrazine, melarsonyl, melarsoprol, melengestrol acetate, meletimide, melinamide, melitracen, melizame, meloxicam, melperone, melphalan, memantine, memotine, menabitan, menadiol, menadiol diphosphate, menadiol disulfate, menadione, menadione sodium bisulfite, menatetrenone, menbutone, menfegol, menglytate, menitrazepam, menoctone, menogaril, menthol, meobentine, meparfynol, mepazine, mepenzolate bromide, meperidine, mephenesin, mephenoxalone, mephentermine, mephenyton, mephobarbital, mepindolol, mepiprazole, mepiroxol, mepitiostane, mepivacaine, mepixanox, mepramidil, meprednisone, meprobamate, meproscillarin, meproxitol, meprylcaine, meptazinol, mequidox, mequinol, mequitazine, meralein, meralluride, merbarone, merbromin, mercaptamine, mercaptomerin, mercaptopurine, mercuderamide, mercufenol chloride, mercumatilin, mercurobutol, mergocriptine, merophan, mersalyl, mesabolone, mesalamine, meseclazone, mesna, mesocarb, meso-hexestrol, mesoridazine, mesipirenone, mestanolone, mesterolone, mestranol, mesudipine, mesulergine, mesulfamide, mesulfen, mesuprine, metabromsalan, metacetamol, metaclazepam, metaglycodol, metahexamide, metamelfalan, metamfazone, metamfepramone, metampicillin, metanixin, metapramine, metaproterenol, metaraminol, metaterol, metaxalone, metazamide, metazide, metazocine, metbufen, meteneprost, metergoline, metergotamine, metescufylline, metesculetol, metethoheptazine, metformin, methacholine chloride, methacycline, methadone, methadyl acetate, methallenestril, methallibure, methalthiazide, methamphetamine, methandriol, methandrostenolone, methaniazide, methantheline bromide, methaphenilene, methapyrilene, m thaqualone, metharbital, methastyridone, methazolamide, methdilazine, methenamine, methenolone acetate, methenolone enanthate, metheptazine, methestrol, methetoin, methicillin, methimazole, methiodal sodium, methioguanine, methiomeprazine, methionine, methisazone, methitural, methixene, methocarbamol, methohexital, methopholine, methoserpidine, methotrexate, methotrimeprazine, methoxamine, methoxsalen, methoxyflurane, methoxyphedrine, methoxyphenamine, methoxypromazine, methscopolamine bromide, methsuximide, methylclothiazide, N-methyladrealone hcl, methyl alcohol, methylatropine nitrate, methylbenactyzium bromide, methylbenzethonium, methylchromone, methyldesorphine, methyldihydromorphine, methyldopa, methyldopate, methylene blue, methylphedrine, methylergonovine, methylformamide, methyl nicotinate, 2-methyl-19-nortestosterone, 2-methyl-11-oxoprogestrone, methyl palmoxirate, methylparaben, methylphendiate, methylprednisolone, methylprednisolone aceponate, methylprednisolone acetate, methylprednisolone hemisuccinate, methylprednisolone phosphate, methylpredn isolone suleptanate, methyl salicylate, methylstreptonigrin, 4-methyltestosterone, 7-methyltestosterone, 17-methyltestosterone, 7-methyltesosterone propionate, methylthionosine, 16-methylthioprogestone, methylthiouracil, methynodiol diacetate, methyprylon, methysergide, metiamide, metiapine, metiazinic acid, metibride, meticrane, metildigoxin, metindizate, metioprim, metioxate, metipirox, metipranolol, metiprenaline, metitepine, metizoline, metkephamid, metochalcone, metocinium iodide, metoclopramide, metocurine iodide, metofenazate, metogest, metolazone, metomidate, metopimazine, metopon, metoprine, metoprolol, metoquizine, metoserpate, metostilenol, metoxepin, metrafazoline, metralindole, metrazifone, metrenperone, metribolone, metrifonate, metrifudil, metrizamide, metrizoic acid, metronidazole, meturedepa, metyrapone, metyridine, metyrosine, mevastatin, mexafylline, mexazolam, mexenone, mexiletine, mexiprostil, mexoprofen, mexrenoate, mezacopride, mezepine, mezilamine, mezlocillin, mianserin, mibolerone, micinicate, miconazole, micronomicin, midaflur, midaglizole, midalcipran, midamaline, midazogrel, midazolam, midecamycin, midodrine, mifentidine, mifepristone, mifobate, miglitol, mikamycin, milacemide, milenperone, milipertine, miloxacin, milrinone, milverine, mimbane, minaprine, minaxolone, mindolilol, mindoperone, minepentate, minocromil, minocycline, minoxidil, mioflazine, mipimazole, mirincamycin, miristalkonium chloride, miroprofen, mirosamicin, misonidazole, misoprostol, mitindomide, mitobronitol, mitoclomine, mitoguazone, mitolactol, mitomycin, mitonafide, mitopodozide, mitoquidone, mitotane, mitotenamine, mitoxantrone, mitozolomide, mivacurium chloride, mixidine, misoprostol, mitindomide, mitobronitol, mitoclomine, mitoguazone, mitolactol, mitomycin, mitonafide, mitopodozide, mitoquidone, mitotane, mitotenamine, mitoxantrone, mitozolomide, mivacurium chloride, mixidine, mizoribine, mobecarb, mobenzoxamine, mocimycin, mociprazine, moclobemide, moctamide, modafinil, modaline, mofebutazone, mofloverine, mofoxime, molfarnate, molinazone, molindone, molracetam, molsidomine, mometasone furoate, monalazone disodium, monensin, monobenzone, monoethanolamine, monometacrine, monophosphothiamine, monothioglycerol, monoxerutin, montirelin, moperone, mopidamol, mopidralazine, moprolol, moquizone, morantel, morazone, morclofone, morforex, moricizine, morinamide, morniflumate, morocromen, moroxydine, morpheridine, morphine, morsuximide, motapizone, motrazepam, motretinide, moveltipril, moxadolen, moxalactam, moxaprindine, moxastine, moxaverine, moxazocine, moxestrol, moxicoumone, moxipraquine, moxisylyte, moxnidazole, moxonidine, mupirocin, murabutide, murocainid muzolimine, mycophenolic acid, myfadol, myralact, myrophine, myrtecaine, nabazenil, nabilone, nabitan, naboctate, nabumetone, nadide, nadolol, nadoxolol, naepaine, nafamostat, nafazatrom, nafcaproic acid, nafcillin, nafenodone, nafenopin, nafetolol, nafimidone, nafiverine, naflocort, nafomine, nafoxadol, nafoxidine, nafronyl, naftalofos, naftazone, naftifine, naftopidil, naftoxate, naftypramide, nalbuphine, nalidixic acid, nalmefene, nalmexone, nalorphine, naltrexone, naminterol, namoxyrate, nanaprocin, nandrolone cyclotate, nandrolone decanoate, nandrolone, phenpropionate, nanofin, nantradol, napactadine, napamezole, naphazoline, naphthonone, naprodoxime, naproxen, naproxol, naranol, narasin, natamycin, naxagolide, naxaprostene, nealbarbital, nebidrazine, nebivolol, nebracetam, nedocromil, nefazodone, neflumozide, nefopam, nelezaprine, neoarsphenamine, neocinchophen, neomycin, neostigmine bromide, nequinate, neraminol, nerbacadol, nesapidil, nesosteine, netilmicin, netobimin, neutramycin, nexeridine, niacin, niacinamide, nialamide, niaprazine, nibroxane, nicafenine, nicainoprol, nicametate, nicarbazin, nicarpidine, nicergoline, niceritrol, niceverine, niclofolan, niclosamide, nicoboxil, nicoclonate, nicocodine, nicocortonide, nicodicodine, nicofibrate, nicofuranose, nicofurate, nicogrelate, nicomol, nicomorphine, nicopholine, nicorandil, nicothiazone, nicotinyl alcohol, nicoxamat, nictiazem, nictindole, nodroxyzone, nifedipine, nifenalol, nifenazone, niflumic acid, nifluridide, nifuradene, nifuraldezone, nifuralide, nifuratel, nifuratrone, nifurdazil, knifurethazone, nifurfoline, nifurimide, nifurizone, nifurmazole, nifurmerone, nifuroquine, nifuroxazide, nifuroxime, nifurpipone, nifurpirinol, nifurprazine, nifurquinazole, nifursemizone, nifursol, nifurthiazole, nifurtimox, nifurtoinol, nifurvidine, nifurzide, niguldipine, nihydrazone, nikethamide, nileprost, nilprazole, niludipine, nilutamide, nilvadipine, nimazone, nimesulide, nimetazepam, nimidane, nimodipine, nimorazole, nimustine, niometacin, niperotidine, nipradilol, niprofazone, niridazole, nisbuterol, nisobamate, nisoldipine, nisoxetine, nisterime acetate, nitarsone, nitazoxanide, nithiamide, nitracrine, nitrafudam, nitralamine, nitramisole, nitraquazone, nitrazepam, nitrefazole, nitrendipine, nitricholine, nitrochlofene, nitrocycline, nitrodan, nitrofurantoin, nitrofurazone, nitroglycerin, nitromersol, nitromide, nitromifene, nitroscanate, nitrosulfathiazole, nitroxinil, nitroxoline, nivazol, nivimeldone, nixylic acid, nizatidine, nizofenone, noberastine, nocloprost, nocodazole, nofecainide, nogalamycin, nolinium bromide, nomegestrol, nomelidine, nomifensine, nonabine, nonaperone, nonapyrimine, nonoxynol-4, nonoxynol-9, noracymethadol, norbolethone, norbudrine, norclostebol, norcodeine, nordazepam, nordefrin, nordinone, norepinephrine, norethandrolone, norethindrone, norethindrone acetate, norethynodrel, noreximide, norfenefrine, norfloxacin, norfloxacin succinil, norflurane, norgesterone, norgestimate, norgestomet, norgestrel, norgestrienone, norletimol, norlevorphanol, normethadone, normethandrone, normorphine, norpipanone, nortestosterone propionate, nortetrazepam, nortriptyline, norvinisterone, nosantine, noscapine, nosiheptide, novobiocin, noxiptiline, noxytiolin, nuclomedone, nuclotixine, nufenoxole, nuvenzepine, nylestriol, nylidrin, nystatin, obidoxime, ociltide, ocrylate, octabenzone, octacaine, octafonium chloride, octamoxin, octamylamine, octanoic acid, octapinol, octastine, octaverine, octazamide, octenidine, octenidine saccharin, octicizer, octimibate, octorylene, octodrine, octopamine, octotiamine, octoxynol-9, octriptyline, octrizole, ofloxacin, ofornine, oftasceine, olaflur, olaquindox, oleanomycin, oletimol, oleyl alcohol, olivomycin a, olmidine, olpimedone, olsalazine, oltipraz, olvanil, omeprazole, omidoline, omoconazole, omonasteine, onapristone, ondansetron, ontianil, opiniazide, opipramol, orazamide, orbutopril, orconazole, orestrate, ormetoprim, ornidazole, ornipressin, ornithine, ornoprostil, orotic acid, orotirelin, orpanoxin, orphenadrine, ortetamine, osalmid, osmadizone, otilonium bromide, otimerate sodium, ouabain, oxabolone cipionate, oxabrexine, oxaceprol, oxacillin, oxadimedine, oxaflozane, oxaflumazine, oxagrelate, oxalinast, oxaliplatin, oxamarin, oxametacin, oxamisole, oxamniquine, oxanamide, oxandrolone, oxantel, oxapadol, oxapium iodide, oxapropanium iodide, oxaprotiline, oxaprozin, oxarbazole, oxatomide, oxazafone, oxazepam, oxazidione, oxazolam, oxazorone, oxcarbazepine, oxdralazine, oxeladin, oxendolone, oxepinac, oxetacillin, oxethazaine, oxetorone, oxfendazole, oxfenicine, oxibendazole, oxibetaine, oxiconazole, oxidopamine, oxidronic acid, oxifentorex, oxifungin, oxilorphan, oximonam, oxindanac, oxiniacic acid, oxiperomide, oxiracetam, oxiramide, oxisopred, oxisuran, oxitefonium bromide, oxitriptan, oxitriptyline, oxitropium bromide, oxmetidine, oxodipine, oxogestone phenpropionate, oxolamine, oxolinic acid, oxomemazine, oxonazine, oxophenarsine, oxoprostol, oxpheneridine, oxprenoate potassium, oxprenolol, oxtriphylline, oxybenzone, oxybutynin, oxychlorosene, oxycinchophen, oxyclozanide, oxycodone, oxydipentonium chloride, oxyfedrine, oxymesterone, oxymetazoline, oxymetholone, oxymorphone, oxypendyl, oxypertine, oxyphenbutazone, oxyphenonium bromide, oxypurinol, oxypyrronium bromide, oxyquinoline, oxyridazine, oxysonium iodide, oxytetracycline, oxytiocin, ozagrel, ozolinone, pacrinolol, pactamycin, padimate, pafenolol, palatrigine, paldimycin, palmidrol, palmoxiric acid, pamabrom, pamaquine, pamatolol, pamidronic acid, pancuronium bromide, panidazole, panomifene, patenicate, panthenol, pantothenic acid, panuramine, papaverine, papaveroline, parachlorophenol, paraflutizide, paraldehyde, paramethadione, paramethasone acetate, paranyline, parapenzolate bromide, parapropamol, pararosaniline, pararosaniline embonate, paraxazone, parbendazole, parconazole, pareptide, parethoxycaine, pargeverine, pargolol, pargyline, paridocaine, parodilol, paromomycin, paroxetine, paroxypropione, parsalmide, partricin, parvaquone, pasiniazid, paulomycin, paxamate, pazelliptine, pazoxide, pcnu, pecilocin, pecocycline, pefloxacin, pelanserin, pelretin, pelrinone, pemedolac, pemerid, pemoline, pempidine, penamecillin, penbutolol, pendecamaine, penfluridol, penflutizide, pengitoxin, penicillamine, penicillin procaine, penicillin, penimepicycline, penimocycline, penirolol, penmesterol, penoctonium bromide, penprostene, pentabamate, pentacynium chloride, pentaerythritol tetranitrate, pentafluranol, pentagastrin, pentagestrone, pentalamide, pentamethonium bromide, pentamethylmelamine, pentamidine, pentamoxane, pentamustine, pentapiperide, pentapiperium methylsulfate, pentaquine, pentazocine, pentetate calcium trisodium, pentetic acid, penthienate bromide, penthrichloral, pentiapine maleate, pentifylline, pentigetide, pentisomicin, pentisomide, pentizidone, pentobarbital, pentolinium tartrate, pentomone, pentopril, pentorex, pentosan polysulfate sodium, pentostatin, pentoxifylline, pentrinitrol, pentylenetrazole, peplomycin, pepstatin, peraclopone, peradoxime, perafensine, peralopride, peraquinsin, perastine, peratizole, perbufylline, perfluamine, perflunafene, pergolide, perhexilene, periciazine, perimetazine, perindopril, perindoprilat, perisoxal, perlapine, permethrin, perphenazine, persilic acid, petrichloral, pexantel, phanquone, phenacaine, phenacemide, phenacetin, phenacttropinium chloride, phenadoxone, phenaglycodol, phenamazoline, phenampromide, phenarsone sulfoxylate, phenazocine, phenazopyridine, phencarbamide, phencyclidine, phendimetrazine, phenelzine, pheneridine, phenesterin, penethicillin, phenformin, phenglutarimide, phenicarbazide, phenindamine, phenindione, pheniprazine, pheniramine, phenisonone, phenmetrazine, phenobarbital, phenobutiodil, phenolphtalein, phenolsulfonphthalein, phenomorphan, phenoperidine, phenothiazine, phenothrin, phenoxybenzamine, phenoxypropazine, phenprobamate, phenprocoumon, phenpromethamine, phensuximide, phentermine, phentolamine, phenylalanine, phenyl aminosalicylate, phenylbutazone, phenylrphrine, phenylethyl alcohol, phenylmercuric acetate, phenylmercuric borate, phenylmercuric chloride, phenylmercuric nitrate, phenylmethylbarbituric acid, phenylpropanolamine, phenylthilone, phenyltoloxamine, phenyramidol, phenytoin, phetharbital, pholcodine, pholedrine, phosphoramide mustard, phoxim, phthalofyne, phthalysulfacetamide, phthalylsulfamethizole, phthalylsulfathiazole, physostigmine, phytic acid, phytonadiol diphosphate, phytonadione, pibecarb, pibenzimol, pibecarb, pibenzimol, piberaline, picafibrate, picartamide, picenadol, picilorex, piclonidine, piclopastine, picloxydine, picobenzide, picodralazine, picolamine, piconol, picoperine, picoprazole, picotamide, picotrin diolamine, picumast, pidolic acid, pifarnine, pifenate, pifexole, piflutixole, pifoxime, piketoprofen, pildralazine, pilocarpine, pimoclone, pimefylline, pimelautde, pimetacin, pimethixene, pimetine, pimetremide, piminodine, pimobendan, pimondiazole, pimozide, pinacidil, pinadoline, pinafide, pinaverium bromide, pinazepam, pincainide, pindolol, pinolcaine, pinoxepin, pioglitazone, pipacycline, pipamazine, pipaperone, pipazethate, pipebuzone, pipecuronium bromide, pipemidic acid, pipenzolate bromid pipequaline, piperacetazine, piperacillin, piperamide, piperazine, piperazinedione, piperidolate, piperilate, piperocaine, piperoxan, piperylone, pipobroman, pipoctanone, pipofezine, piposulfan, pipotiazine palmiate, pipoxizine, pipoxolan, pipradimadol, pipradol, pipramadol, pipratecol, piprinhydrinate, piprocurarium iodide, piprofurol, piprozolin, piquindone, piquizil, piracetam, pirandamine, pirarubicin, piraxelate, pirazmonam, pirazolac, pirbenicillin, pirbuterol, pirdonium bromide, pirenoxine, pirenperone, pirenzepine, pirepolol, piretanide, pirfenidone, piribedil, piridicillin, piridocaine, piridoxilate, piridronic acid, pirifibrate, pirindazole, pirinixic acid, pirinixil, piriprost, piriqualone, pirisudanol, piritramide, piritrexim, pirlimycin, pirlindole, pirmagrel, pirmenol, pirnabine, piroctone, pirogliride, piroheptine, pirolate, pirolazamide, piromidic acid, piroxantrone hcl, piroxicam, piroxicam cinnamate, piroxicillin, piroximone, pirozadil, pirprofen, pirquinozol, pirralkonium bromide, pirtenidine, pitenodil, pitofenone, pituxate, pivampicillin, pivenfrine, pivopril, pivoxazepam, pizotyline, plafibride, plaunotol, pleuromulin, plicamycin, podilfen, podophylloxoxin, poldine methylsulfate, polidocanol, ploymyxin, polythiazide, ponalrestat, ponfibrate, porfiromycin, poskine, potassium guaiacolsulfonate, potassium nitrazepate, potassium sodium tartrate, potassium sorbate, potassium thiocyanate, practolol, prajmalium, pralidoxime chloride, pramipexole, pramiracetam, pramiverine, pramoxime, prampine, pranolium chloride, pranoprofen, pranosal, prasterone, pravastatin, praxadine, prazepam, prazepine, praziquantel, prazitone, prazocillin, prazosin, preclamol, prednazate, prednazoline, prednicarbate, prednimustine, prednisolamate, prednisolone, prednisolone acetate, prednisolone hemisuccinate, prednisolone phosphate, prednisolone steaglate, prednisolone tebutate, prednisone, prednival, pr dnylidene, prefenamate, pregnenolone, pregnenolone succinate, premazepam, prenalterol, prenisteine, prenoverine, prenoxdiazine, prenylamine, pretamazium iodide, pretiadil, pribecaine, pridefine, prideperone, pridinol, prifelone, prifinium bromide, prifuroline, prilocaine, primaperone, primaquine, primidolol, primidone, primycin, prinomide, pristinamycin, prizidilol, proadifen, probarbital, probenecid, probicromil, probucol, procainamide, procaine, procarbazine, procaterol, prochlorperazine, procinolol, procinonide, proclonol, procodazole, procyclidine, procymate, prodeconium bromide, prodilidine, prodipine, prodblic acid, profadol, profexalone, proflavine, proflazepam, progabide, progesterone, proglumetacin, proglumide, proheptazine, proligestone, proline, prolintane, prolonium iodide, promazine, promegestone, promestriqne, promethazine, promolate, promoxolane, pronetalol, propacetamol, propafenone, propamidine, propanidid, propanocaine, propantheline bromide, proparacaine, propatyl nitrate, propazolamide, propendiazole, propentofylline, propenzolate, properidine, propetamide, propetandrol, propicillin, propikacin, propinetidine, propiolactone, propiomazine, propipocaine, propiram, propisergide, propiverine, propizepine, propofol, propoxate, propoxycaine, propoxyphene, propranolol, propyl docetrizoate, propylene glycol, propylene glycol monostearate, propyl gallate, propylhexedrine, propyliodone, propylparaben, propylthiouracil, propyperone, propyphenazone, propyromazine bromide, proquazone, proquinolate, prorenoate potassium, proroxan, proscillaridin, prospidium chloride, prostalene, prosulpride, prosultiamine, proterguride, protheobromine, prothipendyl, prothixene, protiofate, protionamide, protirelin, protizinic acid, protokylol, protoveratine, protriptyline, proxazole, proxibarbal, proxibutene, proxicromil, proxifezone, proxorphan, proxyphylline, prozapine, pseudoephedrine, psilocybine, pumiteba, puromycin, pyrabrom, pyran copolymer, pyrantel, pyrathiazine, pyrazinamide, pyrazofurin, pyricarbate, pyridarone, pyridofylline, pyridostigmine bromide, pyridoxine, pyrilamine, pyrimethamine, pyrimitate, pyrinoline, pyrithione zinc, pyrithyldione, pyritidium bromide, pyritinol, pyronine, pyrophenindane, pyrovalerone, pyroxamine, pyrrobutamine, pyrrocaine, pyrroliphene, pyrrolnitrin, pyrvinium chloride, pytamine, quadazocine, quadrosilan, quatacaine, quazepam, quazinone, quazodine, quazolast, quifenadine, quillifoline, quinacainol, quinacillin, quinacrine, quinaldine blue, quinapril, quinaprilat, quinazosin, quinbolone, quincarbate, quindecamine, quindonium bromide, quindoxin, quinestradol, quinestrol, quinethazone, quinetolate, quinezamide, quinfamide, quingestanol acetate, quingestrpne, quindine, quinine, quinocide, quinpirole, quinterenol, quintiofos, quinuclium bromide, quinupramine, quipazine, quisultazine, racefemine, racemethionine, racemethorphan, racemetirosine, raclopride, ractopamine, rafoxanide, ralitoline, raloxifene, ramciclane, ramefenazone, ramipril, ramiprilat, ramixotidine, ramnodignin, ranimustine, ranimycin, ranitidine, ranolazine, rathyronine, razinodil, razobazam, razoxane, reboxetine, recainam, reclazepam, relomycin, remoxipride, renanolone, rentiapril, repirinast, repromicin, reproterol, recimetol, rescinnamine, reserpine, resorantel, resorcinol, resorcinol monoacetate, retelliptine, retinol, revenast, ribavirin, riboflavin, riboflavin 5′-phosphate, riboprine, ribostamycin, ridazolol, ridiflone, rifabutin, rifamide, rifampin, rifamycin, rifapentine, rifaximin, rilapine, rilmazafone, rilmenidine, rilopirox, rilozarone, rimantadine, rimazolium metilsulfate, rimcazole, rimexolone, rimiterol, rimoprogin, riodipine, rioprostil, ripazepam, risocaine, risperidone, ristianol, ristocetin, ritanserin, ritiometan, ritodrine, ritropirronium bromide, ritrosulfan, robenidine, rocastine, rociverine, rodocaine, rodorubicin, rofelodine, roflurante, rokitamycin, roletamide, rolgamidine, rolicyclidine, rolicyprine, rolipram, rolitetracycline, rolodine, rolziracetam, romifenone, romifidine, ronactolol, ronidazole, ronifibrate, ronipamil, ronnel, ropitoin, ropivacaine, ropizine, roquinimex, rosaprostol, rosaramicin, rosaramicin butyrate, rosaramicin propionate, rosoxacin, rosterolone, rotamicillin, rotoxamine, rotraxate, roxarsone, roxatidine acetate, roxibolone, roxindole, roxithromycin, roxolonium metilsulfate, roxoperonef rufloxacin, rutamycin, rutin, ruvazone, sabeluzole, saccharin, salacetamide, salafibrate, salantel, salazodine, salazossulfadimedine, salazosulfamide, salazosulfathiazole, salethamide, salfluverine, salicin, salicyl alcohol, salicylamide, salicylanilide, salicylic acid, salinazid, salinomycin, salmefanol, salmeterol, salmisteine, salprotoside, salsalate, salverine, sancycline, sangivamycin, saperconazole, sarcolysin, sarmazenil, sarmoxicillin, sarpicillin, saterinone, satranidazole, savoxepin, scarlet red, scopafungin, scopolamine, seclazone, secnidazole, secobarbital, secoverine, securinine, sedecamycin, seganserin, seglitide, selegiline, selenium sulfide, selprazine, sematilide, semustine, sepazonium chloride, seperidol, sequifenadine, serfibrate, sergolexole, serine, sermetacin, serotonin, sertaconazole, sertraline, setastine, setazindol, setiptiline, setoperone, sevitropium mesilate, sevoflurane, sevopramide, siagoside, sibutramine, siccanin, silandrone, silibinin, silicristin, silidianin, silver sulfadiazine, simetride, simfibrate, simtrazene, simvastatin, sinefungin, sintropium bromide, sisomicin, sitalidone, sitofibrate, sitogluside, sodium benzoate, sodium dibunate, sodium ethasulfate, sodium formaldehyde sulfoxylate, sodium gentisate, sodium gualenate, sodium nitrite, sodium nitroprusside, sodium oxybate, sodium phenylacetate, sodium picofosfate, sodium picosulfate, sodium propionate, sodium stibocaptate, sodium stibogluconate, sodium tetradecyl sulfate, sodium thiosulfate, sofalcone, solasulfone, solpecainol, solypertine, somantadine, sopitazine, sopromidine, soquinolol, sorbic acid, sorbinicate, sorbinil, sorbitan monolaurate, sorbitan monooleate, sorbitan monopalmitate, sorbitan monostearate, sorbitan trioleate, sorbitan tristearate, sorbitol, sorndipine, sotalol, soterenol, spaglumic acid, sparfosic acid, sparsomycin, sparteine, spectinomycin, spiclamine, spiclomazine, spiperone, spiradoline, spiramide, spiramycin, spirapril, spiraprilat, spirendolol, spirgetine, spirilene, spirofylline, spirogermanium, spiromustine, spironolactone, spiroplatin, spirorenone, spirotriazine, spiroxasone, spiroxatrine, spiroxepin, spizofurone, stallimycin, stanolone, stanzolol, stearic acid, stearyl alcohol, stearylsulfamide, steffimycin, stenbolone acetate, stepronin, stercuronium iodide, stevaladil, stibamine glucoside, stibophen, stilbamidine, stilbazium iodide, stilonium iodide, stirimazole, stiripentol, stirocainide, stirifos, streptomycin, streptonicozid, streptonigrin, streptovarycin, streptozocin, strinoline, strychnine, styramate, subathizone, subendazole, succimer, succinylcholine chloride, succinylsulfathiazole, succisulfone, suclofenide, sucralfate, sucrose octaacetate, sudexanox, sudoxicam, sufentanil, sufosfamide, sufotidine, sulazepam, sulbactam, sulbactam pivoxil, sulbenicillin, sulbenox, sulbentine, sulbutiamine, sulclamide, sulconazole, sulfabenz, sulfabenzamide, sulfacarbamide, sulfacecole, sulfacetamide, sulfachlorpyridazine, sulfachrysoidine, sulfaclomide, sulfaclorazole, sulfaclozine, sulfacytine, sulfadiazine, sulfadicramide, sulfadimethoxine, sulfadoxine, sulfaethidole, sulfaguandide, sulfaguanole, sulfalene, sulfaloxic acid, sulfamazone, sulfamerazine, sulfameter, sulfamethazine, sulfamethizole, sulfamethoxazole, sulfamethoxypyridazine, sulfamethoxypyridazine acetyl, sulfametomidine, sulfametrole, sulfamonomethoxine, sulfamoxole, sulfanil amide, sulfanitran, sulfaperin, sulfaphenazole, sulfaproxyline, sulfapyridine, sulfaquinoxaline, sulfarsphenamine, sulfasalazine, sulfasomizole, sulfasuccinamide, sulfasymazine, sulfathiazole, sulfathiourea, sulfatolamide, sulfatroxazole, sulfatrozole, sulfazamet, sulfinalol, sulfinpyrazone, sulfiram, sulfisomidine, sulfisoxazole, sulfisoxazole, sulfobromophthalein, sulfonethylmethane, sulfonmethane, sulfonterol, sulforidazine, sulfoxone sodium, sulicrinat, sulindac, sulisatin, sulisobenzone, sulmarin, sulmazole, sulmepride, sulinidazole, sulocarbilate, suloctidil, sulosemide, sulotroban, suloxifen, sulpiride, sulprosal, sulprostone, sultamicillin, sulthiame, sultopride, sultosilic acid, sultroponium, sulverapride, sumacetamol, sumatriptan, sumetizide, sunagrel, suncillin, supidimide, suproclone, suprofen, suramin, suricainide, suriclone, suxemerid, suxethonium chloride, suxibuzone, symclosene, symetine, synephrine, syrisingopine, taclamine, tacrine, taglutimide, talampicillin, talastine, talbutal, taleranol, talinolol, talipexole, talisomycin, talmetacin, talmetoprim, talniflumate, talopram, talosalate, taloximine, talsupram, taltrimide, tameridone, tameticillin, tametraline, tamitinol, tamoxipen, tampramine, tandamine, taprostene, tartaric acid, tasuldine, taurocholic acid, taurolidine, tauromustine, tauroselcholic acid, taurultam, taxol, tazadolene, tazanolast, tazaburate, tazeprofen, tazifylline, taziprinone, tazolol, tebatizole, tebuquine, teclothiazide, teclozan, tedisamil, tefazoline, tefenperate, tefludazine, teflurane, teflutixol, tegafur, telenzepine, temafloxacin, temarotene, temazepam, temefos, temelastine, temocillin, temodox, temozolomide, temurtide, tenamfetamine, tenilapine, teniloxazine, tenilsetam, teniposide, tenocyclidine, tenonitrozole, tenoxicam, tenylidone, teopranitol, teoprolol, tepirindole, tepoxalin, terazosin, terbinafine, terbucromil, terbufibrol, terbuficin, terbuprol, terbutaline, terciprazine, terconazole, terfenadine, terfluranol, terguride, terizidone, ternidazole, terodiline, terofenamate, teroxalene, teroxirone, terpin hydrate, tertatolol, tesicam, tesimide, testolactone, testosterone, testosterone cypionate, testosterone enanthate, testosterone ketolaurate, testosterone phenylacetate, testosterone propionate, tetrabarbital, tetrabenazine, tetracaine, tetrachloroethylene, tetracycline, tetradonium bromide, tetraethylammonium chloride, tetrahydrozoline, tetramethrin, tetramisole, tetrandrine, tetrantoin, tetrazepam, tetriprofen, tetronasin 5930, tetroquinone, tetroxoprim, tetrydamine, texacromil, thalicarpine, thalidomide, thebacon, thebaine, thenalidine, thenium closylate, thenyldiamine, theobromine, theodrenaline, theofibrate, theophylline, thiabendazole, thiacetarsamide, thialbarbital, thiambutosine, thiamine, thiamiprine, thiamphenicol, thiamylal, thiazesim, thiazinamium chloride, thiazolsulforie, thiethyperazine, thihexinol methylbromide, thimerfonate, thimerosal, thiocarbanidin, thiocarzolamide, thiocolchioside, thiofuradene, thioguanine, thioguanine alpha-deoxyriboside, thioguanine beta-deoxyriboside, thioguanosine, thiohexamide, thioinosine, thiopental, thiopropazate, thioproperazine, thioridazine, thiosalan, thiotepa, thiotetrabarbital, thiothixene, thiouracil, thiphenamil, thiphencillin, thiram, thonzonium bromide, thonzylamine, thozalinone, threonine, thymidine, thymol, thymol iodide, thymopentin, thyromedan, thyropropic acid, tiacrilast, tiadenol, tiafibrate, tiamenidine, tiametonium iodide, tiamulin, tianafac, tianeptine, tiapamil, tiapirinol, tiapride, tiaprofenic acid, tiaprost, tiaramide, tiazofurin, tiazuril, tibalosin, tibenalast sodium, tibenzate, tibezonium iodide, tibolone, tibric acid, tibrofan, tic-mustard, ticabesone propionate, ticarbodine, ticarcillin, ticarcillin cresyl, ticlatone, ticlopidine, ticrynafen, tidiacic, tiemoium iodide, tienocarbine, tienopramine, tienoxolol, tifemoxone, tiflamizole, tiflorex, tifluadom, tiflucarbine, tiformin, tifurac, tigemonam, tigestol, tigloidine, tilbroquinol, tiletamine, tilidine, tiliquinol, tilisolol, tilmicosin, tilomisole, tilorone, tilozepine, tilsuprost, timefurone, timegadine, timelotem, timepidium bromide, timiperone, timobesone acetate, timofibrate, timolol, timonacic, timoprazole, tinabinol, tinazoline, tinidazole, tinisulpride, tinofedrine, tinoridine, tiocarlide, tioclomarol, tioconazole, tioctilate, tiodazosin, tiodonium chloride, tiomergine, tiomesterone, tioperidone, tiopinac, tiopronin, tiopropamine, tiospirone, tiotidine, tioxacin, tioxamast, tioxaprofen, tioxidazole, tioxolone, tipentosin, tipepidine, tipetropium bromide, tipindole, tipredane, tiprenolol, tiprinast, tipropidil, tiprostanide, tiprotimod, tiquinamide, tiquizium bromide, tiratricol, tiropramide, tisocromide, tisopurine, tisoquone, tivandizole, tixadil, tixanox, tixocortol pivalate, tizabrin, tianidine, tizolemide, tizoprolic acid, tobramycin, tobuterol, tocainide, tocamphyl, tocofenoxate, tocofibrate, tocophersolan, todralazine, tofenacin, tofetridine, tofisoline, tofisopam, tolamolol, tolazamide, tolazoline, tolboxane, tolbutamide, tolciclate, toldimfos, tolfamide, tolfenamic acid, tolgabide, tolimidone, tolindate, toliodium chloride, toliprolol, tolmesoxide, tolmetin, tolnaftate, tolnapersine, tolnidamine, toloconium metilsulfate, tolonidine, tolonium chloride, toloxatone, toloxychlorinol, tolpadol, tolpentamide, tolperisone, toliprazole, tolpronine, tolpropamine, tolpyrramide, tolquinzole, toirestat, toltrazuril, tolufazepam, tolycaine, tomelukast, tomoglumide, tomoxetine, tomoxiprole, tonazocine, topiramate, toprilidine, tonazocine, topiramate, toprilidine, topterone, toquizine, torasemide, toebafylline, toremifene, tosifen, tosufloxacin, tosulur, toyocamycin, toyomycin, traboxepine, tracazolate, tralonide, tramadol, tramazoline, trandolapril, tranexamic acid, tranilast, transcainide, trantelinium bromide, tranylcypromine, trapencaine, trapidil, traxanox, trazilitine, trazium esilate, trazodone, trazolopride, trebenzomine, trecadrine, treloxinate, trenbolone acetate, trengestone, trenizine, trosulfan, trepibutone, trepipam, trepirium iodide, treptilamine, trequensin, trestolone acetate, trethinium tosilate, trethocanoic acid, tretinoin, tretoquinol, triacetin, triafungin, triamcinolone, triamcinolone acetonide, triamcinolone acetonide-phosphate, triamcinolone benetonide, triamcinolone diacetate, triamcinolone furetonide, triamcinolone hexacetonide, triampyzine, triamterene, triazinate, triaziquone, triazolam, tribendilol, tribenoside, tribromoethanol, tribromsalan, tribuzone, triacetamide, trichlormethiazide, trichlormethine, trichloroacetic acid, trichloroethylene, tricribine phosphate, triclabendazole, triclacetamol, triclazate, triclobisonicum chloride, triclocarban, triclodazol, triclofenol, piperazine, triclofos, triclofylline, triclonide, triclosan, tricyclamol chloride, tridihexethyl chloride, trientine, triethylenemelamine, triethylenephosphoramide, trifenagrel, trifezolac, triflocin, triflubazam, triflumidate, trifluomeprazine, trifluoperazine, trifluperidol, triflupromazine, trifluridine, triflusal, trigevolol, trihexyphenidyl, triletide, trilostane, trimazosin, trimebutine, trimecaine, trimedoxime bromide, trimeperidine, trimeprazine, trimetazidine, trimethadione, trimethamide, trimethaphan camsylate, trimethidinium methosulfate, trimethobenzamide, trimethoprim, trimetozine, trimetrexate, trimexiline, trimipramine, trimoprostil, trimoxamine, trioxifene, trioxsalen, tripamide, triparanol, tripelennamine, tripotassium dicitratobismuthate, triprolidine, tritiozine, tritoqualine, trityl cysteine, trixolane, trizoxime, trocimine, troclosene potassium, trofosfamide, troleandomycin, trolnitrate, tromantadine, tromethamine, tropabazate, tropanserin, tropapride, tropatepine, tropenziline bromide, tropicamide, tropigline, tropiprine, tropodifene, trospectomycin, trospium chloride, troxerutin, troxipide, troxolamide, troxonium tosilate, troxypyrrolium tosilate, troxypyrrolium tosilate, truxicurium iodide, truxipicurium iodide, tryparsamide, tryptophan, tryptophane mustard, tuaminoheptane, tubercidine, tubocurarine chloride, tubulozole, tuclazepam, tulobutrol, tuvatidine, tybamate, tylocrebin, tylosin, tyramine, tyropanic acid, tyrosine, ubenimex, ubidecarenone, ubisindine, ufenamate, ufiprazole, uldazepam, ulobetasol, undecoylium chloride, undecyclenic acid, uracil mustard, urapidil, urea, uredepa, uredofos, urefibrate, urethane, uridine, ursodeoxycholic acid, ursucholic acid, vadocaine, valconazole, valdetamide, valdipromide, valine, valnoctamide, valofane, valperinol, valproate pivoxil, valproic acid, valpromide, valtrate, vancomycin hcl, vaneprim, vanillin, vanitolide, vanyldisulfamide, vapiprost, vecuronium bromide, velnacrine maleate, venlafaxine, veradoline, veralipride, verapamil, verazide, verilopam, verofylline, vesnarinone, vetrabutine, vidarabine, vidarabine phophate, vigabatrin, viloxazine, viminol, vinbarbital, vinblastine, vinburnine, vincamine, vincanol, vincantril, vincofos, vinconate, vincristine, vindrburnol, vindesine, vindepidine, vinformide, vinglycinate, vinorelbine, vinpocetine, vinpoline, vinrosidine, vintiamol, vintriptol, vinylbital, vinylether, vinzolidine, viomycin, viprostol, viqualine, viquidil, virginiamycin factors, viroxime, visnadine, visnafylline, vitamin e, volazocine, warfarin, xamoterol, xanoxic acid, xanthinol niacinate, xanthiol, xantifibrate, xantocillin, xenalipin, xenazoic acid, xenbucin, xenipentone, xenthiorate, xenygloxal, xenyhexenic acid, xenytropium bromide, xibenolol, xibornol, xilobam, ximoprofen, xinidamine, xinomiline, xipamide, xipranolol, xorphanol, xylamidine, xylazine, xylocoumarol, xylometazoline, xyloxemine, yohimbic acid, zabicipril, zacopride, zafuleptine, zaltidine, zapizolam, zaprinast, zardaverine, zenazocine mesylate, zepastine, zeranol, zetidoline, zidapamide, zidometacin, zidovudine, zilantel, zimeldine, zimidoben, zinc acetate, zinc phenolsulfonate, zinc undecylenate, zindotrine, zindoxifene, zinoconazole, zinterol, zinviroxime, zipeprol, zocainone, zofenopril, zoficonazole, zolamine, zolazepam, zolenzepine, zolertine, zolimidine, zoliprofen, zoloperone, zolpidem, zomebazam, zomepirac, zometapine, zonisamide, zopiclone, zorubicin, zotepine, zoxazolamine, zuclomiphene, zuclophenthixol, zylofuramine.
- The following non-limitative examples serve to illustrate the concept of multiple receptor specificity. Other combinations of vectors, spacers and reporters and conjugation technologies leading to multiple vector incorporation are also considered relevant to this invention. Confirmation of the microparticulate nature of products is performed using microscopy as described in WO-A-9607434. Ultrasonic transmission measurements may be made using a broadband transducer to indicate suspensions of products giving an increased sound beam attenuation compared to a standard. Flow dytometric analysis of products can be used to confirm attachment of antibodies thereto. The ability of targeted agents to bind specifically to cells expressing a target may be studied by microscopy and/or using a flow chamber containing immobilised cells, for example employing a population of cells expressing the target structure and a further population of cells not expressing the target. Radioactive, fluorescent or enzyme-labelled streptavidin/avidin may be used to analyse biotin attachment.
- This example is directed at the preparation of targeted microbubbles comprising multiple peptidic vectors arranged in a linear sequence.
a) Synthesis of a Lipopeptide Consisting of a Heparin Sulphate Binding Peptide (KRKR) and Fibronectin Peptide (WOPPRARI).
- The lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Ile-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol, 5% EDT, 5% anisole and 5% H2O for 2 hours giving a crude product yield of 150 mg.
- Purification by preparative HPLC (Vydac 218TP1022 column) of a 40 mg aliquot of crude material was carried out using a gradient of 70 to 100% B over 40 min (A=0.1% TFA/water and B=MeOH) at a flow rate of 9 mL/min. After lyophilization 16 mg of pure material was obtained (Analytical HPLC; Gradient, 70-100% B where B=MeOH, A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 260 and fluorescence, Ex280, Em350—product retention time=19.44 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 2198, found, at 2199.
- b) Preparation of Gas-containing Microbubbles of DSPS ‘Doped’ with a Multiple-specific Lipopeptide Consisting of a Heparin Sulphate Binding Peptide (KRKR) and Fibronectin Peptide (WOPPRARI).
- DSPS (Avanti, 4.5 mg) and lipopeptide from a) (0.5 mg) were weighed into each of 2 vials and 0.8 mL of a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming). The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles rolled overnight. Bubbles were washed several times with deionised water and analysed by Coulter counter (Size: 1-3 micron (87%), 3-5 micron (11.5%)) and acoustic attenuation (frequency max att.: 3.5 MHz). The microbubbles were stable at 120 mm Hg.
- MALDI mass spectral analysis was used to confirm incorporation into DSPS microbubbles as follows; ca. 0.05-0.1 mL of microbubble suspension was transferred to a clean vial and 0.05-0.1 mL methanol added. The suspension was sonicated for 30 s and the solution analysed by MALDI MS. Positive mode gave M+H at 2200, expected for lipopeptide, 2198.
- c) In Vitro Study of Gas-containing Microbubbles of DSPS ‘Doped’ with a Multiple-specific Lipopeptide Consisting of a Heparin Sulphate Binding Peptide (KRKR) and Fibronectin Peptide (WOPPRARI): Binding to Endothelial Cells Under Flow Conditions
- The human endothelial cell line ECV 304, derived from a normal umbilical cord (ATCC CRL-1998) was cultured in 260 mL Nunc culture flasks (chutney 153732) in RPMI 1640 medium (Bio Whittaker) to which L-Glutamine 200 mM, Penicillin/Streptomycin (10.000 U/mL and 10.000 mcg/mL) and 10% Fetal Bovine Serum (Hyclone Lot no. AFE 5183) were added.
- The cells were subcultured with a split ratio of 1:5 to 1:7 when reaching confluence.
- Cover-glasses, 22mm in diameter (BDH, Cat no. 406/0189/40) were sterilised and placed on the bottom of 12 well culture plates (Costar) before cells in 0,5 mL complete medium with serum was added on top.
- When the cells reached confluence the coverslips were placed in a custom made flow-chamber. The chamber consists of a groove carved into a glass plate upon which the cover slip with cells was placed with the cells facing the groove forming a flow channel.
- Ultrasound microbubbles from section b) were passed from a reservoir held at 37 degree Celsius through the flow chamber and back to the reservoir using a peristaltic pump. The flow rate was adjusted to simulate physiological relevant shear rates. The flow chamber was placed under a microscope and the interaction between the microspheres and cells viewed directly. A camera mounted on the microscope was connected to a colour video printer and a monitor.
- A gradual accumulation of the microbubbles on the cells took place which was dependent on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- d) In Vivo Experiment in Dog
- Case 1)
- A 22 kg mongrel dog was anaesthetized with pentobarbital and mechanically ventilated. The chest was opened by a midline sternotomy, the anterior pericardium was removed, and a 30 mm gelled silicone rubber spacer was inserted between the heart and a P5-3 transducer of an ATL HDI-3000 ultrasound scanner. The scanner was set for intermittent short axis imaging once in each end-systole by delayed EGC triggering.
- A net volume of 2 mL of microbubbles from b) were injected as a rapid intravenous bolus. 3 seconds later, the imaged right ventricle was seen to contain contrast material, another 3 seconds later, the left ventricle was also filled, and a transient attenuation shadow that obscured the view of the posterior parts of the left ventricle was observed. A substantial increase in brightness of the myocardium was seen, also in the portions of the heart distal to the left ventricle when the attenuation shadow subsided.
- After passage of the inital bolus, the ultrasound scanner was set to continuous, high frame rate high output power imaging, a procedure known to cause destruction of utrasound contrast agent bubbles in the imaged tissue regions. After a few seconds, the scanner was adjusted back to its initial setting. The myocardium was then darker, and closer to the baseline value. Moving the imaged slice to a new position resulted in re-appearance of contrast effects, moving the slice back to the initial position again resulted in a tissue brightness again close to baseline.
- Case 2) [Comparative]
- A net volume of 2 mL microbubbles prepared in an identical manner to b) above with the exception that no lipopeptide was included in the preparation was injected, using the same imaging procedure as above. The myocardial echo enhancement was far less intense and of shorter duration than observed in case 1. At the completion of the left ventricular attenuation phase, there was also almost complete loss of myocardial contrast effects, and a myocardial echo increases in the posterior part of the left ventricle as in case 1 was not observed.
- This example is directed at the preparation of targeted microbubbles comprising multiple peptidic vectors.
- a) Synthesis of 3-Maleimidopropionylamido-PEG200-acyl Distearoyl Phosphatidylethanolamine (PE-PEG-MAL)
- A mixture of distearoyl phosphatidyl ethanolamine (DSPE), (37.40 mg, 0.005 mmol), N-hydroxysuccinimido-PEG2000-maleimide, NHS-PEG-MAL, (100 mg, 0.25 mmol) and triethylamine (35 μl, 0.25 mmol) in a solution of chloroform/methanol (3:1) was stirred at room temperature for 24 hours. After evaporation of the solvents under reduced pressure, the residue was purified by flash chromatography (chloroform/methanol, 8:2). The product was obtained as a white wax, 92 mg (66%) and structure was verified by NMR and maldi-MS.
- b) Synthesis of RGDC
- The RGDC peptide was synthesised on a ABI 433A automated peptide synthesiser (0.25 mmol scale, Fmoc-Cys(Trt)-Wang resin, (Novabiochem). All amino acids were activated using HBTU. The crude peptide was removed from the resin and simultaneously deprotected in TFA containing 5% EDT, 5% phenol and 5% water. Following evaporation of the excess cleavage solution the peptide was precipitated and triturated several times with diethyl ether before air drying. The crude peptide was purified by preparative hplc and fractions containing pure product combined and freeze dried. Final characterisation was performed using analytical hplc and MALDI MS.
- c) Preparation of Multiple-specific Gas-filled Microbubbles Encapsulated by Phosphatidylserine and ‘Doped’ with RGDC-Mal-PEG3400-DSPE and a Lipopeptide Comprising a Heparin Sulphate Binding Peptide (KRKR) and Fibronectin Peptide (WOPPRARI).
- DSPS (Avanti, 5.0 mg), lipopeptide (0.5 mg) from example 1 a) and PE-PEG-MAL (0.5 mg) from section a) was weighed into a clean vial and 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was sonicated for 3-5 mins, warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter. The mixture was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes. The infranatant was exchanged with 1 mL of PBS containing 1 mg of the peptide RGDC and the pH adjusted to 8. The conjugation reaction was allowed to proceed for 2 h. The bubbles were washed in PBS then with water until all unreacted RGDC had been removed from the infranatant as observed by MALDI-MS. The microbubbles were further analysed by Coulter counter (98% between 1 and 7 micron).
- d) In Vitro Binding Assay.
- The binding of microbubbles to endothelial cells was carried out under flow conditions using the in vitro assay described in example 1. c). A gradual accumulation of the microbubbles on the cells took place which was dependant on the flow rate. Control bubbles not carrying the vectors did not adhere to the endothelial cells detaching from the cells under minimal flow conditions. EXAMPLE 3
- This example is directed at the preparation of microbubbles comprising multiple antibody vectors for targeted ultrasound.
- a) Preparation of Gas-containing Microbubbles Encapsulated with DSPS and PE-PE22000-MAL
- DSPS (Avanti, 4.5 mg) and PE-PEG2000-Maleimide from example 2 a) (0.5 mg) were weighed into a clean vial and 1 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles washed three times with distilled water.
- b) Thiolation of Anti-CD62 and Anti-ICAM-1 Antibodies
- To 0.3 mg each of anti-CD62 and anti-ICAM-1 antibodies dissolved in PBS buffer (pH 7, 0.5 mL) was added Traut's reagent and the solutions stirred at room temperature for 1 h. Excess reagent was separated from the modified protein on a NAP-5 column (Pharmacia).
- c) Conjuaation of Thiolated Anti-CD62 and Anti-ICAM-1 Antibodies to Gas-containing Microbubbles Encapsulated with DSPS and DSPE-PEG2000-MAL
- 0.5 mL of the mixed thiolated antibody preparation from b) was added to an aliquot of microbubbles from a) and the conjugation reaction allowed to proceed for 30 min on a roller table. Following centrifugation at 2000 rpm for 5 min the infranatant was removed. The microbubbles were washed a further three times with water.
- The PEG spacer length may also be varied to include longer e.g. PEG3400 and PEG5000 or shorter e.g. PEG600 or PEG800 chains. Addition of a third antibody such as thiolated-anti-CD34 is also envisaged. EXAMPLE 4
- a) Synthesis of PS Binding/Fibronectin Fragment Fusion Peptide NH2F.N.F.R.L.K.A.G.O.K.I.R.F.G.G.G.G.W.O.P.P.R.A.I.OH.
- The peptide was synthesised on an ABI 433A automatic peptide synthesiser starting with Fmoc-Ile-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids were preactivated using HBTU before coupling.
- The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol, 5% EDT and 5% H2O for 2 hours giving a crude product yield of 302 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 25 mg aliquot of crude material was carried out using a gradient of 20 to 40% B over 40 min (A=0.1% TFA/water and B=0.1% TFA/acetonitrile) at a flow rate of 9 mL/min. After lyophilization 10 mg of pure material was obtained (Analytical HPLC; Gradient, 20 to 50% B where B=0.1% TFA/acetonitrile, A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 214 and 260 nm—product retention time=12.4 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 2856, found, at 2866.
- b) Preparation of Microbubbles of DSPS Coated Non-covalently with Polylysine and the PS Binding/Fibronectin Fragment Fusion Peptide NH2F.N.F.R.L.K.A.G.O.K.I.R.F.G.G.G.G.W.O.P.P.R.A.I.OH.
- DSPS (5 mg, Avanti) was weighed into a clean vial along with poly-L-lysine (Sigma, 0.2 mg) and peptide from a) above (0.2 mg). To the vial was added 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol. The mixture was warmed to 80° C. for 5 minutes. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes.
- Following extensive washing with water, PBS and water the final solution was examined for polylysine and peptide content using MALDI MS. No polypeptide material was observed in the final wash solution.
- Acetonitrile (0.5 mL) was then added and the microbubbles destroyed by sonication. Analysis of the resulting solution for polylysine and PS-binding/fibronectin fusion peptide was then carried out using MALDI MS. The results were as follows:
MALDI expected MALDI found Poly-L-lysine 786, 914, 1042, 1170 790, 919, 1048, 1177 DSPS-binding peptide 2856 2866 - The spacer element contained within the PS binding/Fibronectin fusion peptide (-GGG-) can also be replaced with other spacers such as PEG2000 or poly alanine (-AAA-). It is also envisaged that a form of pre-targeting may be employed, whereby the DSPS binding/Fibronectin fragment fusion peptide is firstly allowed to associate with cells via the fibronectin peptide binding. This is followed by administration of PS microbubbles which then bind to the PS binding peptide.
- This example is directed at the preparation of targeted ultrasound microbubbles whereby streptavidin is used as a linker between biotinylated reporter(s) and vector(s).
- a) Synthesis of Biotin-PEG3400-β-Alanine Cholesterol
- To a solution of cholesteryl-β-alanine hydrochloride (15 mg, 0.03 mmol) in 3 mL chloroform/wet methanol (2.6:1), was added triethylamine (42 mL, 0.30 mmol). The mixture was stired for 10 minutes at room temperature and a solution of biotin-PEG3400-NHS (100 mg, 0.03 mmol) in 1,4-dioxan (1 mL) was added dropwise. After stirring at room temperature for 3 h, the mixture was evaporated to dryness and the residue purified by flash chromatography to give white crystals, yield; 102 mg (89%). The structure was verified by MALDI-MS and NMR.
- b) Synthesis of Biotinylated Endothelin-1 Peptide (Biotin-D-Trp-Leu-Asp-Ile-Ile-Trp.OH)
- The peptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Trp(Boc)-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids were preactivated using HBTU before coupling.
- The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% anisole and 5% H2O for 2 hours giving a crude product yield of 75 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 20 mg aliquot of crude material was carried out using a gradient of 30 to 80% B over 40 min (A=0.1% TFA/water and B=0.1% TFA/acetonitrile) and a flow rate of 9 mL/min. After lyophilization of the pure fractions 2 mg of pure material was obtained (Analytical HPLC; Gradient, 30-80% B where B=0.1% TFA/acetonitrile, A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 214 nm—product retention time=12.6 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 1077, found, 1077.
- c) Synthesis of Biotinyl-fibrin-anti-polymerant Peptide (Biotin-GPRPPERHOS.NH2)
- This peptide was synthesised and purified using similar protocols to those described in section b) above. The pure product was characterised by hplc and MALDI MS.
- d) Preparation of Multiple-specific Gas-filled Microbubbles Encapsulated with Phosphatidylserine and Biotin-PEG3400-β-Alanine Cholesterol
- DSPS (Avanti, 4.5 mg) and biotin-PEG3400-β-Alanine cholesterol from section a) (0.5 mg) were weighed into a vial and 0.8 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming). The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vial was shaken in a cap mixer for 45 s and the microbubbles rolled overnight.
- The microbubble suspension was washed several times with deionised water and analysed by Coulter counter and acoustic attenuation.
- e) Conjugation with Fluorescein Labelled Streptavidin and Biotinylated Peptides from Section b) and c).
- To the microbubble preparation from d) was added fluorescein conjugated streptavidin (0.2 mg) dissolved in PBS (1 mL). The bubbles were placed on a roller table for 3 h at room temperature. Following extensive washing with water and analysis by fluorescence microscopy the microbubbles were incubated in 1 mL of PBS containing biotinyl-Endothelin-1 peptide (0.5 mg) and biotinyl-Fibrin-anti-polymerant peptide (0.5 mg) from sections b) and c) respectively for 2 h. Extensive washing of the microbubbles was performed to remove unconjugated peptide.
- a) Synthesis of Lipopeptide Dipalmitoyl-lysinyl-tryptophanyl-lysinyl-lysinyl-lysinyl(biotinyl)-glycine
- The lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Gly-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol, 5% EDT, 5% anisole and 5% H2O for 2 hours giving a crude product yield of 150 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 40 mg aliquot of crude material was carred out using a gradient of 70 to 100% B over 40 min (A=0.1% TFA/water and B=MeOH) at a flow rate of 9 mL/min. After lyophilization 14 mg of pure material (Analytical HPLC; Gradient, 70-100% B where B=MeOH, A=0.01% TFA/water: column—vydac 218TP54: Detection 13 UV 260 and fluorescence, Ex280, Em350—product retention time=22 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 1478, found, 1471.
- b) Preparation of Gas-containing Microbubbles of DSPS ‘Doped’ with the Biotinylated Lipopeptide Sequence from Section a)
- DSPS (Avanti, 4.5 mg) and lipopeptide from a) (0.5 mg) were weighed into each of 2 vials and 0.8 mL of a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming). The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles formed rolled overnight. The microbubbles were washed several times with deionised water and analysed by Coulter counter and acoustic attenuation.
- MALDI mass spectral analysis was used to confirm incorporation into DSPS microbubbles as described in example 1 b).
- c) Preparation of Multiple-specific Gas-filled Microbubbles Encapsulated with Phosphatidylserine and a Biotinylated Lipopeptide and Functionalised with Streptavidin/biotinyl-endothelin-1 Peptide (Biotin-D-Trp-Leu-Asp-Ile-Ile-Trp.OH)/biotinyl-fibrin-anti-polymerant Peptide (Biotin-GPRPPERHOS.NH2)
- The microbubble preparation from b) above was treated in an analogous manner to that described in example 5 section e).
- a) Preparation of Biotin Containing Microbubbles.
- To a mixture of phosphatidylserine (5 mg, Avanti) and biotin-DPPE (0.6 mg, Pierce) in a clean vial was added 5% propyleneglycol-glycerol in water (1 mL). The dispersion was heated to 80° C. for 5 minutes and then cooled to ambient temperature. The head space was then flushed with perfluorobutane and the vial shaken in a cap-mixer for 45 seconds. After centrifugation the infranatant was removed and the microbubbles formed washed extensively with water.
- b) Conjugation of Gas-filled Microbubbles Encapsulated with Phosphatidylserine and Biotin-DPPE with Streptavidin and a Mixture of Biotinyl-endothelin-1 (Biotin-D-Trp-Leu-Asp-Ile-Ile-Trp.OH) and Biotinyl-Fibrin-anti-polymerant Peptide (Biotin-GPRPPERHOS.NH2)
- The procedure detailed in example 5 section e) was followed.
- a) Synthesis of Succ-PEG3400-DSPE
- NH2-PEG3400-DSPE is carboxylated using succinic anhydride, e.g. by a similar method to that described by Nayar, R. and Schroit, A. J. in Biochemistry (1985) 24, 5967-71.
- b) Preparation of Gas-filled Microbubbles Encapsulated with Phosphatidylserine and Succ-PEG3400-DSPE
- To a mixture (5 mg) of phosphatidylserine (90-99.9 mol %) and Succ-PEG3400-DSPE (10-0.1 mol %) is added 5% propyleneglycol-glycerol in water (1 mL). The dispersion is heated to not more than 80 ° C. for 5 minutes and then cooled to ambient temperature. The dispersion (0.8 mL) is transferred to a vial (1 mL) and the head space is flushed with perfluorobutane. The vial is shaken in a cap-mixer for 45 seconds, whereafter the sample is put on a roller table. After centrifugation the infranatant is exchanged with water and the washing is repeated.
- c) Coupling of Streptavidin to Gas-filled Microbubbles Encapsulated with Phosphatidylserine and Succ-PEG3400-DSPE
- Streptavidin is covalently bound to Succ-PEG3400-DSPE in the membrane by standard coupling methods using a water-soluble carbodiimide. The sample is placed on a roller table during the reaction. After centrifugation the infranatant is exchanged with water and the washing is repeated. The functionality of the attached streptavidin is analysed by binding, e.g. to fluorescently labeled biotin, biotinylated antibodies (detected with a fluorescently labeled secondary antibody) or biotinylated and fluorescence- or radioactively-labeled oligonucleotides. Analysis is performed by fluorescence microscopy or scintillation counting.
- d) Preparation of Multiiple-specific Gas-filled Microbubbles Encapsulated with Phosphatidylserine and Streptavidin-Succ-PEG3400-DSPE Non-covalently Functionalised with Biotinylated Human Transferrin and Human Endothelium IgG Antibody
- Microbubbles from section c) are incubated in a solution containing human transferrin and human endothelium IgG antibody biotinylated using the method described by Bayer et al., Meth. Enzymol., 62, 308. The vector-coated microbubbles are washed as described above.
- a) Synthesis of Succ-PEG3400-DSPE
- Described in example 8 a)
- b) Preparation of Gas-filled Microbubbles Encapsulated with Phosphatidylserine and Succ-PEG3400-DSPE
- Described in example 8b).
- c) Coupling of Streptavidin to Gas-filled Microbubbles Encapsulated with Phosphatidylserine and Succ-PEG3400-DSPE
- Described in example 8 c).
- d) Preparation of Gas-filled Microbubbles Encapsulated with Phosphatidylserine/streptavidin-Succ-PEG-DSPE and the Oligonucleotides Biotin-GAAAGGTAGTGGGGTCGTGTGCCGG and Biotin-GGCGCTGATGATGTTGTTGATTCTT
- Microbubbles from section c) are incubated in a solution containing a mixture of biotin-GAAAGGTAGTGGGGTCGTGTGCCGG and biotin-GGCGCTGATGATGTTGTTGATTCTT. The oligonucleotide-coated microbubbles are washed as described above. Binding of the oligonucleotide to the bubbles is detected e.g. by using fluorescent-labeled oligonucleotides for attachment to the bubbles, or by hybridising the attached oligonucleotide to a labeled (fluorescence or radioactivity) complementary oligonucleotide. The functionality of the oligonucleotide-carrying microbubbles is analysed, e.g. by hybridising the bubbles with immobilized DNA-containing sequences complementary to the attached oligonucleotide.
- Other useful examples include an oligonucleotide complementary to ribosomal DNA (of which there are many copies per haploid genome) and an oligonucleotide complementary to an oncogene (e.g. ras of which there is one copy per haploid genome) are used.
- a) Microbubbles Preparation.
- DSPS (Avanti, 4.5 mg) and DSPE (Avanti, 1.0 mg) were weighed into a clean vial and 1 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vial was shaken in a cap mixer for 45 s and the microbubbles washed two times with distilled water then resuspended in 0.1 M sodium borate buffer pH 9.
- b) Modification of Fibronectin/Transferrin.
- Fibronectin (0.5 mg) and transferrin (1.3 mg) were mixed in PBS and a solution containing NHS-fluorescin in DMSO added. The mixture was stirred at room temperature for 1 hour then the protein purified on a Superdex 200 column. The fluorescein-labelled protein mixture in phosphate buffer pH 7.5 was freeze dried.
- c) Microbubble Modification.
- The freeze-dried product from b) was re-dissolved in 0.5 mL water and to the fluorescein labelled fibronectin/transferrin mixture was added 0.1 mmol of the crosslinker SDBP (Pierce). The solution was incubated on ice for 2 hours, charged on a NAP-5 column and eluted with PBS. To this was added 1 mL of the microbubble suspension from a) and incubation allowed to proceed for 2 h at room temperature on a roller table. Unreacted material was removed by allowing the microbubbles to float then replacing the buffer with water, this process was repeated 3 times.
- This example is directed at the preparation of polymeric ultrasound contrast agents comprising multiple vectors attached to non-surfactant for targeting/therapeutic applications.
- a) Preparation of Polymer Particles Incorporating Avidin in the Polymer Wall
- Hollow polymer particles of P73 (as described in patent WO 96/07434) containing avidin were prepared by a process involving the freeze-drying of an oil-in-water emulsion using the following procedure: An oil solution was prepared by dissolving 0.25 g of the biodegradable polymer P73 [poly(ethylidene bis(16-hydroxyhexadecanoate)co(adipic acid)] in 5 mL of camphene at 60° C. To 0.2 mL of the oil solution was added 2 mg avidin. An aqueous solution was then prepared by dissolving 0.4 g of the polymer, a-(16-hexadecanoyloxyhexadecanoyl)-w-methoxypolyoxyethylene ester, in 20 mL of water at 60° C. The oil solution (0.2 mL) was then mixed with of the aqueous solution (0.8 mL) in a vibromixer (Capmix) for 15 s to form the oil-in-water emulsion. The emulsion was frozen in dry ice and methanol then dried at a pressure of 200 mTorr for 24 h to remove excess solvent. The powder was reconstituted as a suspension of hollow particles by addition of 1.0 mL water. The resulting ultrasound contrast agent was confirmed by microscopy observation, Coulter size distribution, acoustic attenuation and resistance to external pressure.
- b) Synthesis of Biotin-D-Trp-Leu-Asp-Ile-Ile-Trp.OH
- Described in example 5 b).
- c) Conjugation of Polymer Particles Incorporating Avidin.
- The particles from a) were centrifuged and the supernatant replaced with 1 mL of PBS buffer pH 7.5 containing 0.2 mg of biotin-GGCGCTGATGATGTTGTTGATTCTT and 0.2 mg of biotin-D-Trp-Leu-Asp-Ile-Ile-Trp.OH from b) above. After incubation for 24 h the particles were washed extensively with PBS and water.
- a) Preparation of Biotinylated Albumin Microspheres
- A homogeneous suspension of GAM (6×108 particles/mL) in 5 mg/mL albumin was used, with all manipulations being carried out at room temperature. Two 10 mL aliquots were centrifuged (170×g, 5 minutes) to promote floatation of the microspheres and 8 mL of the underlying infranatant was removed by careful suction and replaced by an equal volume of air-saturated phosphate buffered saline, the preparations being rotated for 15-20 minutes to resuspend the microspheres. This procedure was repeated twice, whereafter only negligible amounts of free non-microsphere-associated albumin were assumed to remain. 50 μl of NHS-biotin (10 mM in dimethylsulphoxide) was added to one of the aliquots (final concentration 50 μM); the other (control) aliquot received 50 μl of dimethylsulphoxide. The tubes containing the samples were rotated for 1 hour whereafter 20 μl portions of 50% aqueous glutaraldehyde were added to each tube to crosslink the microspheres. After rotation for another hour the tubes were positioned vertically overnight to allow floatation of the microspheres. The next day, the suspensions were washed twice with phosphate buffered saline containing 1 mg/mL human serum albumin (PBS/HSA) and were resuspended in PBS/HSA after the last centrifugation.
- In order to determine the presence of microsphere-associated biotin, streptavidin conjugated to horseradish peroxidase (strep-HRP) was added to both suspensions and the tubes were rotated for 1 hour to allow for reaction. The microspheres were then washed three times, resuspended in 100 mM citrate-phosphate buffer (pH 5) containing 0.1 mg/mL phenylenediamine dihydrochloride and 0.01% hydrogen peroxide, and rotated for 10 minutes. Development of a yellow-green colour was indicative of the presence of enzyme. The following results were obtained:
Sample Colour development Biotinylated spheres + strp-HRP 2+ Control spheres + strp-HRP + This confirms that GAM were biotinylated.
b) Multiple-specific Gas-containing Microparticles - The biotinylated microspheres are then used to prepare multiple-specific targeting products in an analogous manner to those exemplified in examples 5), 6) and 7).
- a) Synthesis of a Lipopeptide Containing the RGD Sequence and a Fluorescein Reporter Group: Dipalmitoyl-Lys-Lys-Lys-Lys [Acetyl-Arg-Gly-Asp-Lys(Fluorescein)]Gly.OH

- The lipopeptide was synthesised as described in example 1) using commercially available amino acids and polymers. The lipopeptide was cleaved from the resin in TFA containing 5% water, 5% phenol, 5% EDT for 2 h. Following evaporation in vacuo the crude product was precipitated and triturated with diethyl ether. Purification by preparative HPLC (Vydac 218TP1022 column) of a 40 mg aliquot of crude material was carried out using a gradient of 60 to 100% B over 40 min (A=0.1% TFA/water and B=0.1% TFA/acetonitrile) at a flow rate of 9 mL/min. After lyophilization 10 mg of pure material (Analytical HPLC; Gradient, 60-100% B where B=0.1% TFA/acetonitrile), A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 260—product retention time=20-22 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 1922, found, at 1920.
- b) Synthesis of a Lipopeptide Containing a Heparin Sulphate Binding Sequence and a Fibronectin Peptide
- Synthesis and purification described in example 1 a).
- c) Preparation of Multiiple-specific Gas-containing Microbubbles of DSPS Functionalised with a Heparin Sulphate Binding Peptide a Fibronectin Peptide Acetyl-RGD Peptide and Fluorescein.
- DSPS (Avanti, 4 mg) and lipopeptide from a) (0.5 mg, 0.2 mmol) and lipopeptide from b) (0.5 mg) were weighed into each of 2 vials and 0.8 mL of a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming). The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles formed rolled overnight. The microbubbles were washed several times with deionised water and analysed by MALDI mass spectrometry as described in example 1 b). The microbubbles following analysis by microscopy were seen to consist of a range of sizes between 1 and 5 micron. Furthermore the microbubbles were fluorescent.
- This example is directed at the preparation of multiple vector targeted ultrasound agents.
- a) Synthesis of an Endothelial Cell Binding Lipopeptide: 2-n-hexadecylstearyl-Lys-Leu-Ala-Leu-Lys-Leu-Ala-Leu-Lys-Ala-Leu-Lys-Ala-Ala-Leu-Lys-Leu-Ala-NH2.
- The lipopeptide shown below was synthesised on a ABI 433A automatic peptide synthesiser starting with a Rink amide resin on a 0.1 mmol scale using 1 mmol amino acid cartridges.

- All amino acids and 2-n-hexadecylstearic acid were preactivated using HBTU before coupling. The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% EDT, and 5% H2O for 2 hours giving a crude product yield of 150 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 40 mg aliquot of crude material was carried out using a gradient of 90 to 100% B over 50 min (A=0.1% TFA/water and B=MeOH) at a flow rate of 9 mL/min. After lyophilization 10 mg of pure material was obtained (Analytical HPLC; Gradient, 90-100% B where B=MeOH, A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 214 nm—product retention time=23 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 2369, found, at 2373.
- b) Preparation of Gas-containing Microbubbles of DSPS ‘Doped’ with a Endothelial Cell Binding Lipopeptide and PE-PEG2000-MAL
- DSPS (Avanti, 4.5 mg) and lipopeptide from a) (0.5 mg) along with PE-PEG2000-Maleimide from example 2 (0.5 mg) were weighed into a clean vial and 1 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles washed three times with distilled water.
- c) Thiolation of FITC-labelled Anti-transferrin Receptor Antibody.
- FITC labelled CD71 anti-transferrin receptor Ab (100 mg/mL, Becton Dickinson), 0.7 mL, in PBS was modified with Traut's reagent (0.9 mg, Pierce) at room temperature for 1 h. Excess reagent was separated from modified protein on a NAP-5 column (Pharmacia).
- d) Conjugation of Thiolated FITC-labelled Anti-transferrin Receptor Antibody to Gas-containing Microbubbles of DSPS ‘Doped’ with an Endothelial Cell Binding Lipopeptide and DSPE-PEG2000-MAL
- A 0.5 mL aliquot of the protein fraction (2 mL in total) from c) above was added to the microbubbles from b) and the conjugation reaction allowed to proceed for 10 min on a roller table. Following centrifugation at 1000 rpm for 3 min the protein solution was removed and the conjugation repeated a further two times with 1 mL and 0.5 mL aliquots of protein solution respectively. The bubbles were then washed four times in distilled water and a sample analysed for the presence of antibody by flow cytometry and microscopy. A fluorescent population of >92% was observed.

- Incorporation into the microbubbles of lipopeptide was confirmed by MALDI mass spectrometry as described in example 1 b).
- This example is directed to the preparation of microbubbles containing multiple protein vectors for targeted ultrasound/therapy.
a) Synthesis of a Thiol Functionalised Lipid Molecule: Dipalmitoyl-Lys-Lys-Lys-Aca-Cys.OH
- The lipid structure shown above was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Cys(Trt)-Wang resin (Novabiochem) on a 0.25 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU coupling chemistry.
- The simultaneous removal of peptide from the resin and deprotection of side-chain protecting groups was carried out in TFA containing 5% EDT, and 5% H2O for 2 hours giving a crude product yield of 250 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 40 mg aliquot of crude material was carried out using a gradient of 90 to 100% B over 50 min (A=0.1% TFA/water and B=MeOH) at a flow rate of 9 mL/min. After lyophilization 24 mg of pure material was obtained (Analytical HPLC; Gradient, 70-100% B where B=0.1% TFA/acetonitrile, A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 214 nm-product retention time=23 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 1096, found, at 1099.
- b) Preparation of Gas-containing Microbubbles of DSPS ‘Doped’ with a Thiol Containing Lipid Structure:
- DSPS (Avanti, 4.5 mg) and the lipid structure from a) above (0.5 mg) were weighed into a clean vial and 0.8 mL of a solution containing 1.4% propylene glycol/2.4% glycerol in water added. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming) and filtered while still hot through a 40 micron filter. The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles placed on roller table overnight. Bubbles were washed several times with deionised water and analysed for thiol group incorporation using Ellmans Reagent.
- c) Modification of Transferrin and Avidin with Fluorescein-NHS and Sulpho-SMPB.
- To a mixture of 2 mg of transferrin (Holo, human, Alpha Therapeutic Corp) and 2 mg of avidin (Sigma) in PBS (1 mL) was added 0.5 mL DMSO solution containing 1 mg Sulpho-SMPB (Pierce) and 0.5 mg Fluorescein-NHS (Pierce). The mixture was stirred for 45 minutes at room temperature then passed through a Sephadex 200 column using PBS as eluent. The protein fraction was collected and stored at 4° C. prior to use.
- d) Microbubble Conjugation with Modified Transferrin/Avidin.
- To the thiol containing microbubbles from b) was added 1 mL of the modified transferrin/avidin protein solution c). After adjusting the pH of the solution to 9 the conjugation reaction was allowed to proceed for 2 h at room temperature. Following extensive washing with deionised water the microbubbles were analysed by Coulter counter (81% between 1 and 7 micron) and fluorescence microscopy (highly fluorescent microbubbles were observed).
- This example is directed to the preparation of microbubbles having a reactive group on the surface for non-specific targeting, principally utilising disulphide exchange reactions to effect binding to a multiplicity of cellular targets.
- DSPS (Avanti, 5.0 mg) and the thiol containing lipid structure from example 15 a)(1.0 mg) were weighed into a clean vial and 0.8 mL of a solution containing 1.4% propylene glycol/2.4% glycerol in water added. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming) and filtered while still hot through a 40 micron filter. The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles placed on roller table overnight. Bubbles were washed several times with deionised water and analysed for thiol group incorporation using Ellmans Reagent.
- This example is directed to the preparation of ultrasound agents for combined targeting and therapeutic applications.
a) Synthesis of a Lipopeptide Functionalised with Captopril: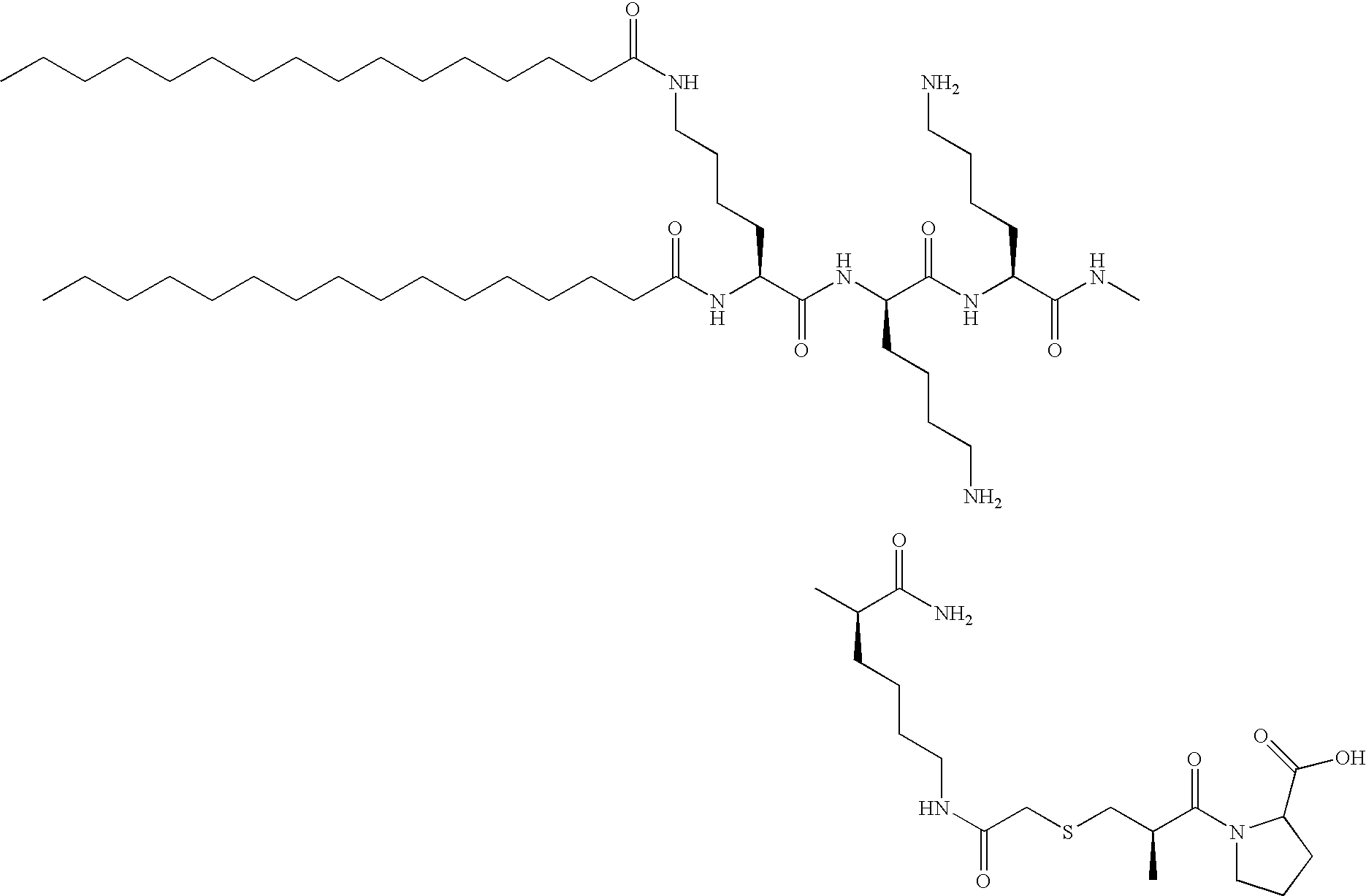
- The structure shown above was synthesised using a manual nitrogen bubbler apparatus starting with Fmoc protected Rink Amide MBHA resin (Novabiochem) on a 0.125 mmol scale. All amino acids were purchased from Novabiochem and palmitic acid from Fluka. Coupling was carried out using standard TBTU/HOBt/DIEA protocols. Bromoacetic acid was coupled through the side-chain of Lys as a symmetrical anhydride using DIC preactivation. Captopril (Sigma) dissolved in DMF was introduced on the solid-phase using DBU as base.
- Simultaneous removal of the peptide from the resin and deprotection of side-chain protecting groups was carried out in TFA containing 5% EDT, 5% water and 5% ethyl methyl sulphide for 2 h. An aliquot of 10 mg of the crude material was purified by preparative liquid chromatography (Vydac 218TP1022 column) using a gradient of 70 to 100% B over 60 min (A=0.1% TFA/water and B=0.1%
- TFA/acetonitrile) at a flow rate of 10 mL/min. After lyophilization a yield of 2 mg of pure material was obtained (analytical HPLC: gradient 70-100% B over 20 min, A=0.1% TFA/water and B=0.1% TFA/acetonitrile; flow rate 1 mL/min; column Vydac 218TP54; detection UV 214 nm; retention time 26 min). Further characterisation was carried out using MALDI mass spectrometry, giving M+H at 1265 as expected.
b) Synthesis of a Lipopeptide with Affinity for Endothelial Cells: Dipalmitoyl-Lys-Lys-Lys-Aca-Ile-Arg-Arg-Val-Ala-Arg-Pro-Pro-Leu-NH2
- The lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Rink amide resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol, 5% EDT and 5% H2O for 2 hours giving a crude product yield of 160 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 35 mg aliquot of crude material was carried out using a gradient of 70 to 100% B over 40 min (A=0.1%
- TFA/water and B=MeOH) at a flow rate of 9 mL/min. After lyophilization 20 mg of pure material was obtained (Analytical HPLC; Gradient, 70-100% B where B=MeOH, A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 214 and 260 nm—product retention time=16 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 2050, found, at 2055.
- c) Preparation of Gas-containing Microbubbles of DSPS Comprising a Lipopeptide for Endothelial Cell Targeting and a Captopril Containing Molecule for Drug Delivery
- DSPS (Avanti, 4.5 mg), product from a) (0.5 mg) and product from b) (0.5 mg) were weighed into a vial and 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol was added to each vial. The mixture was warmed to 80° C. for 5 minutes (vials shaken during warming). The samples were cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were firstly shaken in a cap mixer for 45 s then rolled for 1 h followed by extensive washing with deionised water. No detectable levels of starting material were found in the final wash solution as evidenced by MALDI MS.
- MALDI mass spectral analysis was used to confirm incorporation of the products from section a) and b) into the microbubbles as described in example 1 b).
- d) In Vitro Study of Gas-containing Microbubbles of DSPS Comprising a Lipopepitde for Endothelial Cell Targeting and a Captopril Containing Molecule for Therapeutic Applications.
- The in vitro assay described in example 1 c) was used to examine cell binding under flow conditions. A gradual accumulation of the microbubbles on the cells took place which was dependant on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- This example is directed at the preparation of targeted microbubbles comprising multiple peptidic vectors having a combined targeting and a therapeutic application.
- a) Synthesis of a Lipopeptide Comprising a Helical Peptide with Affinity for Cell Membranes:hexadecylstearyl-Lys-Leu-Ala-Leu-Lys-Leu-Ala-Leu-Lys-Ala-Leu-Lys-Ala-Ala-Leu-Lys-Leu-Ala-NH2.
- Described in example 14 a).
- b)—Preparation of Multiiple-specific Gas-containing Microbubbles.
- DSPS (Avanti, 5.0 mg), lipopeptide from a)(0.3 mg)and polymixin B sulphate (Sigma, 0.5 mg) were weighed into a clean vial and 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was sonicated for 3-5 mins, warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter. The mixture was cooled to room temperature and the head space flushed with perfluorobutane gas. The vial was shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes. The microbubbles were washed in water until no polymixin B sulphate or lipopeptide could be detected in the infranatant by MALDI-MS. Microscopy showed that the size distribution of the bubble population was between 1-8 micron as desired.
- To the washed bubbles (ca. 0.2 mL) was added methanol (0.5 mL) and the mixture placed in a sonic bath for 2 min. The resulting clear solution, following analysis by MALDI-MS, was found to contain both lipopeptide and polymixin B sulphate (expected 1203, found 1207).
- This example is directed at the preparation of targeted microbubbles comprising multiple vectors for targeted/therapeutic/drug release applications.
a) Synthesis of a Lipopeptide Comprising an Interleukin-1 Receptor Binding Peptide: Dipalmitoyl-Lys-Gly-Asp-Trp-Asp-Gln-Phe-Gly-Leu-Trp-Ara-Gly-Ala-Ala.OH
- The lipopeptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Ala-Wang resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- The simultaneous removal of lipopeptide from the resin and side-chain protecting groups was carried out in TFA containing 5% H20, 5% anisole, 5% phenol and 5% EDT for 2 hours giving a crude product yield of 150 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 30 mg aliquot of crude material was carried out using a gradient of 90 to 100% B over 40 min (A=0.1% TFA/water and B=MeOH) at a flow rate of 9 mL/min. After lyophilization 4 mg of pure material was obtained (Analytical HPLC; Gradient, 90-100% B over 20 min where B=MeOH, A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 214 nm; product retention time=23 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 2083, found, at 2088.
b) Synthesis of a Branched Methotrexate Core Structure Containing a Thiol Moiety.
- The methotrexate structure was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Cys(Trt) Tentagel resin on a 0.1 mmol scale. The simultaneous removal of product from the resin and deprotection of protecting groups was carried out in TFA containing 5% EDT and 5% H2O for 2 hours giving a crude product yield of 160 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 30 mg aliquot of crude material was carried out using a gradient of 10 to 30% B over 40 min (A=0.1% TFA/water and B=0.1% TFA/acetonitrile) and a flow rate of 9 mL/min. After lyophilization of the pure fractions 9 mg of pure material was obtained (Analytical HPLC; Gradient, 5-50% B where B=0.1% TFA/acetonitrile, A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 214 nm—product retention time=9.5 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 1523, found, 1523.
- c)—Preparation of Multiiple-specific Gas-containing Microbubbles.
- DSPS (Avanti, 4.5 mg.) and thiol containing lipopeptide from example 15 a) (0.5 mg) and lipopeptide from a) (0.2 mg) above were weighed into a clean vial and 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol added. The mixture was sonicated for 3-5 mins, warmed to 80° C. for 5 minutes then filtered through a 4.5 micron filter. The mixture was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles centrifuged at 1000 rpm for 3 minutes following which the infranatant was discarded.
- d) Conjugation of Methotrexate Branched Structure to Thiolated Microbubbles.
- The methotrexate structure from b) above (0.5 mg) was dissolved in PBS pH 8.0. The solution was then added to the thiol containing bubbles from c) and disulphide bond formation allowed to proceed for 16 h. Following extensive washing with PBS and water the bubbles were analysed by microscopy and MALDI MS.
- It is also considered relevant that the disulphide bond linking the methotrexate structure to the microbubble may be reduced in vivo liberating the free drug molecule. This in combination with a tumour specific vector is a drug delivery system. A physiologically relevant reducing agent such as glutathione may be used to bring about drug release.
- This example is directed to the preparation of microbubbles for gene therapy/anti-sense applications. It is envisaged that specific targeting may be achieved by further doping of microbubble membranes with vector modified lipid structures as described in example 1.
- a) Preparation of DSPS Gas-containing Microbubbles
- DSPS (Avanti, 4.5 mg) was weighed into a clean vial. 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol was added and the mixture sonicated for 2 min then warmed to 80° C. for 5 minutes. Immediately following warming the solution was filtered through a 4 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vial was shaken in a cap mixer for 45 s. Bubbles were then washed once with deionised water and the infranatant discarded. The microbubbles were then resuspended in 0.5 mL water.
- b) Preparation of Poly-L-lysine/DNA Complex and Loading of DSPS Microbubbles
- To 1 mg of poly-L-lysine (70-150 kD) in a clean vial was added 0.1 mL of a fluorescein labeled digest of plasmid pBR322 (Biorad) dissolved in TE buffer (10 mM tris-HCl, pH 8). The solution was made up to a total of 0.6 mL by addition of water and the pH adjusted to 8.
- Complexation was allowed to proceed for 1 h then 0.05 mL of the polylysine-DNA solution was added to the microbubble suspension from a) above. After. 1 h microscopy was used to show that the bubbles were fluorescent confirming the presence of DNA.
- This example is directed to the preparation of microbubbles having a thiol group on the surface for modification with thiol containing vectors for targeting/drug delivery and drug release.
a) Synthesis of the Branched Peptide Dabsyl-Tyr-Arg-Ala-Leu-Val-Asp-Thr-leu-Lys-Lys (NH2-Arg-Gly-Asp-Ser)-Gly-Cys.OH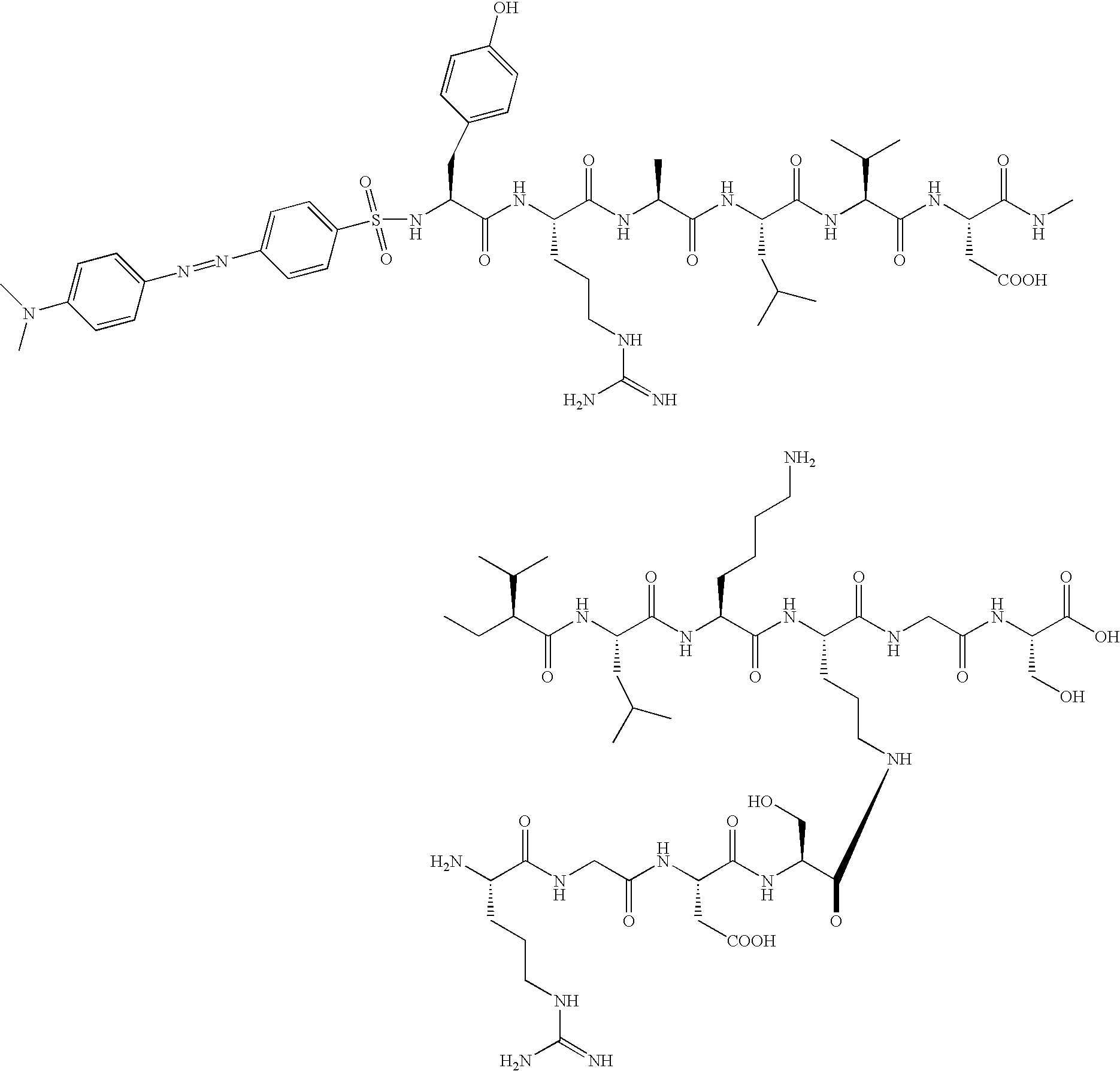
- The peptide was synthesised on a ABI 433A automatic peptide synthesiser starting with Fmoc-Cys(Trt)-Tentagel resin on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids were preactivated using HBTU before coupling.
- The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol, 5% EDT and 5% H2O for 2 hours giving a crude product yield of 160 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 30 mg aliquot of crude material was carried out using a gradient of 10 to 60% B over 40 min (where A=0.1% TFA/water and B=acetonitrile) at a flow rate of 9 mL/min. After lyophilization 2.5 mg of pure material was obtained (Analytical HPLC; Gradient, 10-50% B over 20 min where B=0.1% TFA/acetonitrile and A=0.01% TFA/water: column—vydac 218TP54: Detection—UV 214 and 435 nm—product retention time=21 min). Further product characterization was carried out using MALDI mass spectrometry; expected, M+H at 2070, found, at 2073.
- b) Preparation of Thiol Containing Gas-filled Microbubbles.
- As described in example 15 a) and b).
- c) Oxidative Coupling of Thiolated Microbubbles with Multiple-specific Peptide Via Disulphide Bond Formation.
- The infranatant from the microbubbles from b) above was discarded and replaced with a solution of dabsyl-peptide from a) (1 mg) in 0.7 mL dil. ammonia solution (pH 8). To this was added 0.2 mL of a stock solution containing 6 mg of potassiumferricyanate dissolved in 2 mL of water. The vial was placed on a roller table and-thiol oxidation allowed to proceed for 2 h. The bubbles were then washed extensively with water until the infranatant was free of the dabsyl-peptide as evidenced by hplc and MALDI MS.
- Detection of microbubble bound peptide was carried out by reduction of the disulphide bond using the water souble reducing agent tris-(2-carboxyethyl)-phosphine. Following reduction the infranatant was found to contain free dabsyl-peptide as evidenced by hplc and MALDI MS.
- Other physiological relevant reducing agents such as reduced glutathione are also considered useful for initiating release.
- a) Synthesis of Z-Ala-polymer (3-O-(carbobenzyloxy-L-alanyl)-polymer)
- The polymer is prepared from ethylidene bis(16-hydroxyhexadecanoate) and adipoyl chloride as described in WO-A-9607434, and a polymer fraction with molecular weight 10000 is purified using gel permeation chromatography (GPC). 10 g of the material (corresponding to 1 mmol OH groups), Z-alanine (5 mmol) and dimethylaminopyridine (4 mmol) are dissolved in dry dimethylformamide/tetrahydrofuran and dicyclohexylcarbodiimide is then added. The reaction mixture is stirred at ambient temperature overnight. Dicyclohexylurea is filtered off and the solvent is removed using rotary evaporation. The product is purified by chromatography, fractions containing the title compound are combined and the solvent is removed using rotary evaporation. The structure of the product is confirmed by NMR.
- b) Synthesis of Ala-polymer (3-O-(L-alanyl)-polymer)
- Z-Ala-polymer (0.1 mmol) is stirred in toluene/tetrahydrofuran and glacial acetic acid (15% of the total volume) and hydrogenated in the presence of 5% palladium on charcoal for 2 hours. The reaction mixture is filtered and concentrated in vacuo.
- c) Synthesis of Biotinamidocaproate-Ala-polymer
- A solution of Biotinamidocaproate N-hydroxysuccinimide ester in tetrahydrofuran is added to H2N-Ala-polymer dissolved in a mixture of tetrahydrofuran and dimethylformamide and 0.1 M sodium phosphate buffer having a pH of 7.5. The reaction mixture is heated to 30° C. and stirred vigorously; the reaction is followed by TLC to completion. The solvent is evaporated and the crude product is used without further purification.
- d) Gas-containing Particles Comprising Biotin-amidocaproate-Ala-polymer and PEG 10000 Methyl Ether 16-hexadecanoyloxyhexadecanoate
- 10 mL of a 5% w/w solution of biotin-amidocaproate-Ala-polymer in (−)-camphene maintained at 60° C. is added to 30 mL of an 1% w/w aqueous solution of PEG 10000 methyl ether 16-hexadecanoyloxyhexadecanoate (prepared as described in WO-A-9607434) at the same temperature. The mixture is emulsified using a rotor stator mixer (Ultra Turax® T25) at a slow speed for several minutes, and thereafter is frozen in a dry ice/methanol bath and lyophilized for 48 hours, giving the title product as a white powder.
- e) Acoustic Characterisation and Microscopy of the Product
- Confirmation of the microparticulate nature of the product is performed using light microscopy as described in WO-A-9607434. Ultrasonic transmission measurements using a 3.5 MHz broadband transducer indicate that a particle suspension of <2 mg/mL gives a sound beam attenuation of at least 5 dB/cm.
- f) Multiple-specific Microparticles
- The biotinylated microspheres are then used to prepare multiple-specific targeting products similar to those exemplified in examples 5), 6) and 7).
- a) Synthesis of Biotin-PEG3400-acvl-phosphatidyl Ethanolamine
- A mixture of dipalmitoyl phosphatidyl ethanolamine, (21.00 mg, 0.03 mmol), biotin-PEG-CO2—NHS, (100 mg, 0.03 mmol) and triethylamine (42 μl, 0.30 mmol) in a solution of chloroform/methanol (3:1) was stirred at room temperature for 2 hours. After evaporation of the solvents under reduced pressure, the residue was flash chromatographed (methylene chloride/methanol/water, 40:8:1). The product was obtained as a yellow gum, 112 mg (94%) and structure verified by NMR and MALDI-MS.
- b) Binding of Fluorescein-conjugated Streptavidin to Gas Filled Microbubbles
- Gas-containing microbubbles were prepared by mixing DSPS and biotin-PEG3400-acyl-phosphatidyl ethanolamine as described in example 5 a).
- The microbubble suspension was divided into 0.2 mL aliquots and fluorescein conjugated streptavidin added as shown in the table below. The samples were incubated on a roller table for 15 or 30 minutes at ambient temperature before removal of excess protein by washing in PBS.
- Results:
Added Particle Streptavidin Incubation % Fluo- median Aliquot (μg/200: 1 time (amb. rescent diameter no. sample) temp.) particles (microns) 1 0 2.0 — 2 0 — 12 (foam) 3 0.2 30 min 7.8 3.9 (3 × 10−9 mmol) 4 2 30 min 26.2 4.2 (3 × 10−8 mmol) 5 10 15 min 30.5 na (1.5 × 10−7 mmol) 6 20 30 min 97.9 5.2 (3 × 10−7 mmol) 7 40 15 min 96.7 5.1 (6 × 10−7 mmol) 8 DSPS 20 15 min 0.6 3.7 control (3 × 10−7 mmol) - The samples were analysed by flow cytometry and Coulter Counter. The results are summarized in the table above.
- c) Conjugation of Streptavin Coated Microbubbles with the Oligonucleotide Biotin-GAAAGGTAGTGGGGTCGTGTGCCGG and Biotinylated Fibrin-anti-polymerant Peptide Biotin-GPRPPERHOS
- The particles from aliquot no. 6 above were centrifuged and the supernatant replaced with 1 mL of PBS buffer pH 7.5 containing 0.2 mg of biotin-GAAAGGTAGTGGGGTCGTGTGCCGG and 0.2 mg of biotin-GPRPPERHQS (example 5 c). After incubation for 24 h the particles were washed extensively with PBS and water.
- It is envisaged that other biotinylated vectors or therapeutic agents may be conjugated to streptavidin or avidin coated microbubbles using this procedure.
- This example is directed at the preparation of thrombus targeted US agents comprising a therapeutic thromolytic agent.
a) Synthesis of a Lipopeptide with Affinity for Thrombi (Diplamitoyl-Lys-Asn-Gly-Asp-Phe-Glu-Glu-Ile-Pro-Glu-Glu-Tyr-Leu-Gln.NH2).
- The lipopeptide was synthesised on a ABI 433 A automatic peptide synthesiser starting with Rink amide resin (Novabiochem) on a 0.1 mmol scale using 1 mmol amino acid cartridges. All amino acids and palmitic acid were preactivated using HBTU before coupling.
- The simultaneous removal of peptide from the resin and side-chain protecting groups was carried out in TFA containing 5% phenol, 5% EDT, 5% anisole and 5% H2O for 2 h giving a crude product yield of 80 mg. Purification by preparative HPLC (Vydac 218TP1022 column) of a 20 mg aliquot of the crude material was carried out. After lyophilization 6 mg of pure material was obtained. The product was characterized by MALDI mass spectrometry and analytical HPLC.
- b) Modification of Tissue Plasminogen Activator with Sulpho-SMPB.
- A solution of 0.1 mL of ammonium carbonate buffer containing 0.1 mg of t-PA (Sigma) was made up to 0.2 mL by the addition of water. To this solution was added 0.4 mg of Sulpho-SMPB (Pierce) dissolved in 0.05 mL DMSO. The protein solution was left standing at room temperature for 45 min then purification carried out on a Superdex 200 column. The product was eluted in PBS and the modified protein fraction collected.
- c) Preparation of Microbubbles Encapsulated with DSPS/thrombi-binding Lipopeptide and Thiol Containing Lipoeptide and Conjugation to Modified Tissue Plasminogen Activator.
- DSPS (Avanti, 5.0 mg) was weighed into a clean vial along with 0.5 mg of the lipopeptide from a) and 0.5 mg of the thiol containing lipopeptide from example 15 a). To this was added 1.0 mL of a solution of 1.4% propylene glycol/2.4% glycerol and the mixture sonicated for 2 min then warmed to 80° C. for 5 minutes. Immediately following warming the solution was filtered through a 4 micron filter. The sample was cooled to room temperature and the head space flushed with perfluorobutane gas. The vials were shaken in a cap mixer for 45 s and the microbubbles washed 2 times with deionised water. The infranatant was discarded and replaced with a 1 mL aliquot of the protein solution from b) above. The conjugation reaction was allowed to proceed for 1 h. The bubbles were centrifuged and infranatant exchanged with a further 1 mL of protein solution. The incubation step was repeated until all protein solution was used up. The microbubbles were then washed extensively with water and analysed by Coulter counter. The microbubbles were tested in the flow chamber assay described in example 1 c). Microbubbles modified with protein were found to bind in higher numbers than those comprising either lipopeptide/DSPS or DSPS alone.
- It is envisaged that the targeting/therapeutic/ultrasound activities of these microbubbles be evaluated in models of in vitro and in vivo thrombogenisis.
- a) Synthesis of a Lipopeptide Consisting of a Heparin Sulphate Binding Peptide (KRKR) and a Fibronectin Peptide (WOPPRARI)
- Synthesis and purification described in example 1 a).
- b) Synthesis of a Protected Atenolol Derivative Suitable for Solid Phase Coupling
- i) Synthesis of methyl 4-[(2,3-epoxy)propoxy]phenylacetate
- A mixture of methyl 4-hydroxyphenylacetate (4.98 g, 0.03 mol), epichlorohydrin (23.5 ml, 0.30 mol) and pyridine (121 μl, 1.5 mmol) was stirred at 85° C. for 2 h. The reaction mixture was cooled, and excess epichlorohydrin was distilled off (rotavapor). The residue was taken up in ethyl acetate, washed with brine and dried (Na2SO4). The solution was filtered and concentrated. The dark residue was chromatographed (silica, hexane/ethyl acetate 7:3) to give 2.25 g (34%) of a colourless oil. 1H (300 MHz) and 13C NMR (75 MHz) spectra were in accordance with the structure.
- ii) Synthesis of methyl 4- [2-hydroxy-3-[(1-methylethyl)amino]-propoxy]phenylacetate
- A mixture of methyl 4-[(2,3-epoxy)propoxy]phenylacetate (2.00 g, 9.00 mmol), isopropylamine (23 ml, 0.27 mol) and water (1.35 ml, 74.7 mmol) was stirred at room temperature overnight. The reaction mixture was concentrated (rotavapor) and the oily residue was dissolved in chloroform and dried (Na2SO4). Filtration and concentration gave quantitative yield of a yellow oil that was used in the next step without further purification. The structure was verified by 1H and 13C NMR analysis.
- iii) Synthesis of 4-[2-hydroxy-3-[(1-methylethyl)amino]-propoxy]phenylacetic acid hydrochloride
- A solution of methyl 4-[2-hydroxy-3-[(1-methylethyl)amino]-propoxy]phenylacetate (563 mg, 2.00 mmol) in 6 M hydrochloric acid (15 ml) was heated at 100 ° C. for 4 h. The reaction mixture was concentrated (rotavapor) and the residue was taken up in water and lyophilised. 1H and 13C NMR spectra were in accordance with the strucure and MALDI mass spectrometry gave a M+H at 268 as expected.
- iv) Synthesis of N-Boc-4-[2-hydroxy-3-[(1-methylethyl)amino]-propoxy]phenylacetic acid
- A solution of the 4-[2-hydroxy-3-[(1-methylethyl)amino]-propoxy]phenylacetic acid hydrochloride (2.0 mmol) in water (2 ml) was added to a solution of sodium bicarbonate (0.60 g, 7.2 mmol) in water/dioxane (2:1, 15 ml). A solution of di-tert-butyl dicarbonate (0.48 g, 2.2 mmol) in dioxane (5 ml) was added. Progress of the reaction was monitored by TLC analysis (silica, CHCl3/MeOH/AcOH 85:10:5), and portions of di-tert-butyl dicarbonate were added until conversion was complete.
- The reaction mixture was poured onto water saturated with potassium hydrogensulphate and organic material was extracted into ethyl acetate. The organic phase was washed with water and brine, dried (Na2SO4) and filtered to give 0.6 g of crude material. The product was purified by chromatography (silica, CHCl3/MeOH/AcOH 85:10:5). The solution was concentrated and the residue was taken up in glacial acetic acid and lyophilised. Yield 415 mg (56%), white solid. The structure was confirmed by 1H and 13C NMR analysis.
c) Synthesis of a Lipopeptide Functionalised with Atenolol
- The structure shown above was synthesised on a manual nitrogen bubbler starting with Fmoc protected Rink Amide MBHA resin (Novabiochem) on a 0.125 mmol scale, using amino acids from Novabiochem, palmitic acid from Fluka and the compound from a). Coupling was carried out using standard TBTU/HOBt/DIEA protocols.
- Simultaneous removal of the peptide from the resin and deprotection of side-chain protecting groups was carried out in TFA containing 5% EDT and 5% water for 2 h. Crude material was precipitated from ether and purified by preparative liquid chromatography (Vydac 218TP1022 column) using a gradient of 70 to 100% B over 60 min (A=0.1% TFA/water and B=0.1% TFA/acetonitrile) at a flow rate of 10 ml/min. After lyophilisation a yield of 38 mg of pure material was obtained (analytical HPLC: gradient 70-100% B over 20 min, A=0.1% TFA/water and B=0.1% TFA/acetonitrile, flow rate 1 ml/min, column Vydac 218TP54, detection UV 214 nm, retention time 25 min). Further characterisation was carried out using MALDI mass spectrometry (ACH matrix), giving M+H at 1258, expected 1257.
- d) Preparation of Gas-filled Microbubbles of DSPS Comprising a Lipopeptide Cosisting of a Heparin Sulphate Binding Peptide (KRKR) and a Fibronectin Peptide (WOPPRARI) and a Lipopeptide Containing Atenolol
- A solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5.0 mg), product from a) (0.5 mg) and product from c) (0.5 mg) in a vial. The mixture was sonicated for 5 min and then heated at 80 ° C. for 5 min (vial was shaken during warming). The solution was filtered and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water.
- Incorporation of atenolol containing lipopeptide into the bubbles was confirmed by MALDI-MS as described in example 1 b).
- e) In Vitro Study of Multiiple-specific Gas-filled Microbubbles.
- In vitro analysis of the microbubble suspension was carried out as described in example 1 c). A gradual accumulation of the microbubbles on the cells took place which was dependent on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- This example is directed at non-specific modification of a multiplicity of cell receptors on endothelial cells.
- a) Synthesis of Cholesterol 4-[4-[bis(2-chloroethyl)amino]-phenyl]butanoate
- DIC (170 μl, 1.10 mmol) was added to a solution of chlorambucil (Sigma, 669 mg, 2.20 mmol) in dry dichloromethane (15 ml). The mixture was stirred at room temperature for 0.5 h and added to a solution of cholesterol (Aldrich, 387 mg, 1.00 mmol) and DMAP (122 mg, 1.00 mmol) in dichloromethane (10 ml). The reaction mixture was stirred overnight and then poured onto 5% sodium bicarbonate. The phases were separated and the organic phase was washed with brine and dried (MgSO4). The solution was filtered and concentrated and the product was purified by column chromatography (silica, chloroform) to give 560 mg (83%) yield of colouless oil. The product was characterised by MALDI mass spectrometry, giving M+H at 674 as expected. Further characterisation was carried out using 1H (500 MHz) and 13C (125 MHz) NMR analysis, giving spectra in accordance with the structure.
- b) Preparation of Gas-containing Microbubbles of DSPS Comprising a Cholesteryl Ester of Chlorambucil for Diagnostic and/or Therapeutic Applications
- A solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 4.5 mg) and product from a) (0.5 mg) in a vial. The mixture was sonicated for 5 min and then heated at 80 ° C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water. MALDI mass spectrometry showed no detectable level of compound from a) in the final wash solution. Incorporation of chlorambucil cholesteryl ester into the bubbles was confirmed by MALDI-MS as follows: ca 50 μl of microbubbles were transferred to a clean vial containing ca 100 μl of 90% methanol. The mixture was sonicated for 30 s and analysed by MALDI-MS, giving a M+H peak at 668 corresponding to structure from a).
- In combination with a tumour specific vector these microbubbles are considered useful as targeted drug delivery agents.
- a) Synthesis of a Protected Atenolol Derivative Suitable for Solid Phase Coupling
- As described in example 25 section b).
- b) Synthesis of a Lipopeptide Functionalised with Atenolol
- As described in example 25 section c).
- c) Synthesis of Cholesteryl 4-[4-[bis(2-chloroethyl)amino]phenyl]butanoate
- As described in example 25 section d).
- d) Preparation of Microbubbles of DSPS Comprising a Lipopeptide Containing Atenolol and a Cholesteryl Ester of Chloambucil
- A solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5.0 mg), product from b) (0.5 mg) and c) (0.5 mg) and in a vial. The mixture was sonicated for 5 min and then warmed to 80° C. for 5 min (vial was shaken during warming). The solution was filtered and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water. Incorporation of atenolol containing lipopeptide and chlorabucil analogue into the bubble membrane was confirmed by MALDI-MS as described in example 1 c).
- e) In Vitro Study of Multiiple-specific PFB Gas-containing Microbubbles of DSPS Comprising a Lipopeptide Containing Atenolol and a Cholesterol Derivative of Chlorambucil for Diagnostic and Therapeutic Applications
- The in vitro assay described in example 1 c) was used to assess cellular binding under flow conditions. A gradual accumulation of the microbubbles on the cells took place which was dependant on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- a) Synthesis of a Protected Atenolol Derivative Suitable for Solid Phase Coupling
- As described in example 25 section b).
- b) Synthesis of a Lipopeptide Functionalised with Atenolol
- As described in example 25 section c).
- c) Synthesis of Cholanic Acid Thiol Ester of Captopril
- A mixture of 5-β-cholanic acid (Sigma, 361 mg, 1.00 mmol) and DIC (77 μl, 0.50 mmol) in dichloromethane (5 ml) was stirred for 10 min and then added to a solution of captopril (Sigma, 130 mg, 0.600 mmol) and DBU (180 μl, 1.20 mmol) in dichloromethane (10 ml). The reaction mixture was stirred overnight and then poured onto dilute hydrochloric acid. Chloroform (30 ml) was added. The phases were separated and the organic phase was washed with water and brine and dried (MgSO4). After filtration and concentration the crude material was chromatographed (silica, chloroform/methanol/acetic acid 95:4:1). The product was lyophilised from a acetonitrile/water/ethanol mixture. Yield 137 mg (49%) of off-white solid. The structure was verified by 1H (500 MHz) and 13C (125 MHz) NMR spectroscopy. Further characterisation was carried out using MALDI mass spectrometry, giving a M+Na peak in positive mode at m/z 584.
- d) Preparation of Gas-filled Microbubbles of DSPS Comprising a Lipopeptide Containing Atenolol for Cell Targeting and a Lipophilic Thiol Ester of Captopril for Therapeutic Use.
- A solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5.0 mg) and product from b) (0.5 mg) and c) (0.5 mg) in a vial. The mixture was sonicated for 5 min and then heated at 80 ° C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water. MALDI mass spectrometry showed no detectable level of compound from b) and c) in the final wash solution. Incorporation of compounds from b) and from c) into the bubbles was confirmed by MALDI-MS as follows. Ca. 50 μl of microbubbles were transferred to a clean vial containing ca 100 μl of 90% methanol. The mixture was sonicated for 30 s and analysed by MALDI-MS (ACH-matrix),giving peaks according to structures from b) and c), respectively.
- e) In Vitro Study of Gas-containing Microbubbles from d)
- The in vitro assay described in example 1 c) was used to assess cellular binding under flow conditions. A gradual accumulation of the microbubbles on the cells took place which was dependent on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- a) Synthesis of Cholesterol N-Boc-β-alaninate
- DIC (510 μl) was added to a solution of Boc-β-Ala-OH (1.25 g, 6.60 mmol) in dichloromethane (15 ml) under an inert atmosphere. The reaction mixture was stirred for 30 min and then transferred to a flask containing a solution of cholesterol (1.16 g, 3.00 mmol) and DMAP (367 mg, 3.00 mmol) in dichloromethane (15 ml). The reaction mixture was stirred for 2 h and then mixed with an aqueous solution of potassium hydrogensulphate. The phases were separated and the aqueous phase extracted with chloroform. The combined organic phases were washed with aqueous potassium hydrogensulphate and water and dried over MgSO4. After filtration and evaporation the crude product was chromatographed (silica, chloroform/methanol 99:1) to give 1.63 g (97%) of white solid. The structure was confirmed by 1H NMR (500 MHz).
- b) Synthesis of Cholesterol β-alaninate Hydrochloride
- A solution of compound from a) (279 mg, 0.500 mmol) in 1 M hydrochloric acid in 1,4-dioxan (5 ml) was stirred at room temperature for 4 h. The reaction mixture was concentrated to give a quantitative yield of cholesteryl β-alaninate hydrochloride. The structure was confirmed by 1H NMR (500 MHz) analysis and by MALDI mass spectrometry, giving a M+Na peak at 482, expected 481.
- c) Biotin-PEG3400-β-Ala-Cholesterol
- To a solution of cholesteryl β-alaninate hydrochloride (15 mg, 0.03 mmol) in chloroform/wet methanol (2.6:1, 3 ml) was added triethylamine (42 μl, 0.30 mmol). The mixture was stirred for 10 minutes at room temperature and a solution of biotin-PEG3400-NHS (100 mg, 0.03 mmol) in 1,4-dioxane (1 ml) was added dropwise. After stirring at room temperature for 3 hours, the mixture was evapourated to dryness and the residue purified by flash chromatography to give white crystals, yield; 102 mg (89%). The structure was verified by MALDI-MS and by NMR analysis.
- d) Synthesis of Cholesterol 4-[4-[bis(2-chloroethyl)amino]phenyl]butanoate
- DIC (170 μl, 1.10 mmol) was added to a solution of chlorambucil (Sigma, 669 mg, 2.20 mmol) in dry dichloromethane (15 ml). The mixture was stirred at room temperature for 0.5 h and added to a solution of cholesterol (Aldrich, 387 mg, 1.00 mmol) and DMAP (122 mg, 1.00 mmol) in dichloromethane (10 ml). The reaction mixture was stirred overnight then poured into a solution of 5% sodium bicarbonate. The organic phase was washed with brine and dried over MgSO4. The solution was filtered and concentrated and the product was purified by column chromatography (silica, chloroform) to give 560 mg (83%) yield of colouless oil. The product was characterised by MALDI mass spectrometry, giving M+H at 674 as expected. Further characterisation was carried out using 1H (500 MHz) and 13C (125 MHz) NMR analysis, giving spectra in accordance with the structure.
- e) Preparation of Gas-filled Microbubbles
- A solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 5 mg) and product from c) (0.5 mg) and d) (0.5 mg) in a vial. The mixture was sonicated for 5 min and then heated at 80° C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water. MALDI mass spectrometry showed no detectable level of compound from c and d) in the final wash solution.
- Incorporation of compounds from c) and d) into the bubbles was confirmed by MALDI-MS as described in example 1 b).
- This example is directed at the preparation of functionalised microbubbles with non-specific affinity for a multiplicity of cell surface molecules.
a) Synthesis of a Lipopeptide Containing Chlorambucil
- The structure shown above was synthesised on a manual nitrogen bubbler starting with Fmoc protected Rink Amide MBHA resin (Novabiochem) on a 0.125 mmol scale. Standard amino acids were purchased from Novabiochem and palmitic acid from Fluka. Coupling was carried out using standard TBTU/HOBt/DIEA protocol. Chlorambucil (Sigma) was coupled through the side-chain of Lys as a symmetrical anhydride using DIC preactivation.
- Simultaneous removal of the peptide from the resin and deprotection of side-chain protecting groups was carried out in TFA containing 5% EDT, 5% water and 5% ethyl methyl sulphide for 2 h. An aliqout of 10 mg of the crude material was purified by preparative liquid chromatography (Vydac 218TP1022 column) using a gradient of 70 to 100% B over 60 min (A=0.1% TFA/water and B 0.1% TFA/acetonitrile) at a flow rate of 10 ml/min. After lyophilisation a yield of 30 mg of pure material was obtained (analytical HPLC: gradient 70-100% B over 20 min, A=0.1% TFA/water and B=0.1%
- TFA/acetonitrile; flow rate 1 ml/min; column Vydac 218TP54; detection UV 214 nm; retention time 26.5 min). Further characterisation was carried out using MALDI mass spectrometry, giving M+H at 1295, expected 1294.
- b) Preparation of Gas-filled Microbubbles Comprising a Lipopeptide Containing Chlorambucil for Diagnostic and Therapeutic Applications
- A solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 4.5 mg) and product from a) (0.5 mg) in a vial. The mixture was sonicated for 5 min and then heated at 80 ° C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water. MALDI mass spectrometry showed no detectable level of compound from a) in the final wash solution.
- Incorporation of chlorambucil containing lipopeptide into the bubbles was confirmed by MALDI-MS as follows. Ca 50 μl of microbubbles were transferred to a clean vial containing ca 100 μl of 90% methanol. The mixture was sonicated for 30 s and analysed by MALDI-MS (ACH-matrix), giving a M+H peak at 1300, expected at 1294-and a M+Na peak at 1324, expected 1317.
- c) In Vitro Study of Gas-containing Microbubbles of DSPS ‘Doped’ with a Lipopeptide Containing Chlorambucil for Diagnostic and Therapeutic Applications
- The microbubbles were evaluated using the in vitro flow assay described in example 1 c). A gradual accumulation of the microbubbles on the cells took place which was dependant on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- a) Synthesis of a Protected Atenolol Derivative Suitable for Solid Phase Coupling
- As described in example 25) b)
- b) Synthesis of N-[(S)-3-hexadecylthio-2-methylpropionyl]proline
- DIEA (188 μl, 1.10 mmol) was added to a solution of 1-iodohexadecane (176 mg, 0.500 mmol), captopril (120 mg, 0.550 mmol) and DBU (165 μl, 1.10 mmol) in tetrahydrofuran (5 ml). The mixture was heated at 70° C. for 2 h and then concentrated. The residue was poured onto water saturated with potassium hydrogensulphate and organic material was extracted into chloroform. The organic phase was washed with water and dried (MgSO4). The product purified by chromatography (silica, CHCl3/MeOH/AcOH 85:10:5) and lyophilised to give 105 mg (48%) of white solid material. The structure was verified by 1H (500 MHz) and 13C (125 MHz) analysis and further characterised by MALDI mass spectrometry, giving M−H in negative mode at m/z 440 as expected.
- c) Preparation of Gas-filled Microbubbles of DSPS Comprising a Lipopeptide Containing Atenolol and a Lipophilic Derivative of Captopril for Diagnostic and Therapeutic Applications
- A solution of 1.4% propylene glycol/2.4% glycerol (1.0 ml) was added to a mixture of DSPS (Avanti, 4.5 mg), product from b) (0.5 mg) and c) in a vial. The mixture was sonicated for 5 min and then heated at 80°0 C. for 5 min (vial was shaken during warming) and cooled. Head space was flushed with perfluorobutane gas and the vial was shaken in a cap mixer for 45 s followed by extensive washing with deionised water. MALDI mass spectrometry showed no detectable level of compound from b)or c) in the final wash solution. Incorporation of compound b) and c) containing lipopeptide into the bubbles was confirmed by MALDI-MS as described in example 1 b).
- d) In Vitro Study of Gas-containing Microbubbles of DSPS Comprising a Lipopeptide Containing Atenolol and a Lipophilic Derivative of Captopril for Diagnostic and Therapeutic Applications
- The microbubbles were evaluated using the in vitro flow assay described in example 1 c). A gradual accumulation of the microbubbles on the cells took place which was dependant on the flow rate. By increasing the flow rate the cells started to become detached from the coverslip, the microbubbles were still bound to the cells. Control bubbles not carrying the vector did not adhere to the endothelial cells and disappeared from the cells under minimal flow conditions.
- This example was carried out to show that the invention could also be used for cell separation.
- The human endothelial cell line ECV 304, derived from a normal umbilical cord (ATCC CRL-1998) was cultures in Nunc culture flasks (chutney 153732) in RPMI 1640 medium (Bio Whitaker) to which L-Glutamine 200 mM, Penicillin/Streptomycin (10.000 U/ml and 10.00 mcg/ml) and 10% Fetal Calf Serum (Hyclone Lot no AFE 5183) were added. The cells were subcultured following trypsination with a split ratio of 1:5 to 1:7 when reaching confluence. 2 mill. cells from trypsinated confluent cultures were added to a set of 5 centrifuge tubes followed by either control microbubbles of DSPS, microbubbles from example 1 or microbubbles of DSPS doped with the endothelial cell binding lipopeptide from example 14 a) at a concentration of 2, 4, 6, 8 or 10 mill bubbles per tube. The cells at the bottom of the tubes after centrifugation at 400 g for 5 minutes were counted by Coulter counter. It was found that binding of four or more microbubbles to a cell brought about floatation. Furthermore all cells were floated by the endothelial cell binding lipopeptide bubbles while around 50% were floated with microbubbles from example 1).
- This example is directed at the preparation of targeted microbubbles for gene transfer.
- a) Preparation of DSPS Lipopeptide Bubbles/PFB Gas Coated with Polyl-L-lysine
- DSPS (4,5 mg) and lipopeptide from 17 b) (0.5 mg) were weighed in two 2-ml vials. To each vial, 0.8 ml propyleneglycol/glycerol (4%) in water was added. The solution was heated at 80° C. for 5 minutes and shaken. The solution was then cooled to ambient temperature and the headspace flushed with perfluorobutane. The vials were shaken on a Capmix (Espe Capmix, 4450 oscillations/min) for 45 seconds and put on a roller table for 5 minutes. The content of the vials were mixed and the sample washed by centrifugation at 2000 rpm for 5 minutes. The infranatant was removed and the same volume of distilled water added. The washing procedure was repeated once.
- poly-L-lysine HBr (Sigma, 20.6 mg) was dissolved in 2 mL water then an aliquot (0.4 mL) made up to 2 mL water. To 1.2 mL of the diluted poly-L-lysine solution was added 0.12 mL of the DSPS-lipopeptide bubble suspension. Following incubation excess polylysine was removed by extensive washing with water.
- b) Transfection of Cells
- Endothelial cells (ECV 304) were cultured in 6 well plates to a uniform subconfluent layer. A transfection mixture consisting of 5 μg DNA (an Enhanced Green Fluorescent Protein vector from CLONTECH) and 50 μl of microbubble suspension from a) in RPMI medium at a final volume of 250 μl was prepared. The mixture was left standing for 15 min at room temperature then 1 mL of complete RPMI medium added. The medium was removed from the cell culture dish, and the DNA-microbubble mixture added to the cells.
- The cells were incubated in a cell culture incubator (37° C.).
- c) Ultrasonic Treatment
- After 15 minutes incubation, selected wells were exposed to continious wave ultrasound of 1 MHz, 0.5 W/cm2, for 30 seconds.
- d) Incubation and Examination
- The cells were further incubated in the cell culture incubator (37 ° C.) for approximately 4½ hours. The medium containing DNA-microbubbles was then removed by aspiration, and 2 ml complete RPMI medium was added. The cells were incubated for 40-70 hours before examination. Most of the medium was then removed, and the cells were examined by fluorescence microscopy. The results were compared to the results from control experiments were DNA or DNA-polylysine were added to the cells.
Claims (38)
1. A targetable diagnostic and/or therapeutically active agent comprising a suspension in an aqueous carrier liquid of a reporter comprising gas-containing or gas-generating material, said agent being capable of forming at least two types of binding pairs with a target.
2. An agent as claimed in claim 1 wherein the gas comprises air, nitrogen, oxygen, carbon dioxide, hydrogen, an inert gas, a sulphur fluoride, selenium, hexafluoride, a low molecular weight hydrocarbon, a ketone, an ester, a halogenated low molecular weight hydrocarbon or a mixture of any of the foregoing.
3. An agent as claimed in claim 2 wherein the gas comprises a perfluorinated ketone, perfluorinated ether or perfluorocarbon.
4. An agent as claimed in claim 2 wherein the gas comprises sulphur hexafluoride or a perfluoropropane, perfluorobutane or perfluoropentane.
5. An agent as claimed in claim 1 comprising gas microbubbles stabilised by a coalescence resistant surface membrane, a filmogenic protein, a polymer material, a non-polymeric and non-polymerisable wall-forming material or a surfactant.
6. An agent as claimed in claim 5 wherein said surfactant comprises at least one phospholipid.
7. An agent as claimed in claim 6 wherein at least 750 of the said surfactant material comprises phospholipid molecules individually bearing net overall charge.
8. An agent as claimed in claim 7 wherein at least 750 of the film-forming surfactant material comprises one or more phospholipids selected from phosphatidylserines, phosphatidylglycerols, phosphatidylinositols, phosphatidic acids and cardiolipins.
9. An agent as claimed in claim 8 wherein at least 800 of said phospholipids comprise phosphatidylserines.
10. An agent as claimed in claim 1 wherein said gas-containing or gas-generating material is conjugated to at least two vectors or to one vector capable of binding to at least two binding sites.
11. An agent as claimed in claim 1 wherein said gas-containing or gas-generating material is conjugated to one or more targeting vectors having specificity for one or more cellular surface receptors and further comprises moieties capable of binding to a receptor system so as to induce a therapeutic response.
12. An agent as claimed in claim 1 wherein the vector or vectors are selected from antibodies; cell adhesion molecules; cell adhesion molecule receptors; cytokines; growth factors; peptide hormones and pieces thereof; non-bioactive binders of receptors for cell adhesion molecules, cytokines, growth factors and peptide hormones; oligonucleotides and modified oligonucleotides; DNA-binding drugs; protease substrates/inhibitors; molecules generated from combinatorial libraries; small bioactive molecules; and proteins and peptides which bind to cell-surface proteoglycans.
13. An agent as claimed in claim 1 wherein the vector or vectors have affinity for targets at a level such that the agent interacts with but does not fixedly bind to said targets.
14. An agent as claimed in claim 13 wherein the vector or vectors are selected from ligands for cell adhesion proteins and cell adhesion proteins which have corresponding ligands on endothelial cell surfaces.
15. An agent as claimed in claim 1 wherein the vector or vectors are sited such that they are not readily exposed to the target.
16. An agent as claimed in claim 1 wherein the vector or vectors are coupled or linked to the reporter by means of avidin-biotin and/or streptavidin-biotin interactions.
17. An agent as claimed in claim 1 wherein the vector or vectors may be covalently or non-covalently coupled or linked to the reporter.
18. An agent as claimed in claim 1 wherein the vector is coupled or linked to the reporter by means of electrostatic charge interaction.
19. An agent as claimed in claim 1 which further contains moieties which are radioactive or are effective as X-ray contrast agents, light imaging probes or spin labels.
20. An agent as claimed in claim 1 further comprising a therapeutic compound.
21. An agent as claimed in claim 20 wherein said therapeutic compound is an antineoplastic agent, blood product, biological response modifier, antifungal agent, hormone or hormone analogue, vitamin, enzyme, antiallergic agent, tissue factor inhibitor, platelet inhibitor, coagulation protein target inhibitor, fibrin formation inhibitor, fibrinolysis promoter, antiangiogenic, circulatory drug, metabolic potentiator, antitubercular, antiviral, vasodilator, antibiotic, antiinflammatory, antiprotozoan, antirheumatic, narcotic, opiate, cardiac glycoside, neuromuscular blocker, sedative, local anaesthetic, general anaesthetic or genetic material.
22. An agent as claimed in claim 20 wherein said therapeutic compound is covalently coupled or linked to the reporter through disulphide groups.
23. An agent as claimed in claim 20 wherein a lipophilic or lipophilically-derivatised therapeutic compound is linked to the reporter through hydrophobic interactions.
24. A combined formulation comprising:
i) a first administrable composition comprising a pre-targeting vector having affinity for a selected target; and
ii) a second administrable composition comprising an agent as claimed in any of the preceding claims, said agent comprising a vector having affinity for said pre-targeting vector.
25. A combined formulation as claimed in claim 24 wherein said pre-targeting vector comprises a monoclonal antibody.
26. A combined formulation comprising:
i) a first administrable composition comprising an agent an agent as claimed in claim 1 , and
ii) a second administrable composition comprising a substance capable of displacing or releasing said agent from its target.
27. A combined formulation comprising:
i) a first administrable composition comprising an agent as claimed in claim 22 , and
ii) a second administrable composition comprising a reducing agent capable of reductively cleaving the disulphide groups coupling or linking the therapeutic compound and reporter in the agent of said first administrable composition.
28. A process for the preparation of a targetable diagnostic and/or therapeutically active agent as defined in claim 1 which comprises coupling or linking at least one vector to a reporter comprising gas-containing or gas-generating material: such that said agent is capable of forming at least two types of binding pairs with a target.
29. A process as claimed in claim 28 wherein a therapeutic compound is also combined with the reporter.
30. (canceled).
31. A method of generating enhanced images of a human or non-human animal body which comprises administering to said body an agent as claimed in claim 1 and generating an ultrasound, magnetic resonance, X-ray, radiographic or light image of at least a part of said body.
32. A method as claimed in claim 31 which comprises the steps:
i) administering to said body a pre-targeting vector having affinity for a selected target; and thereafter
ii) administering an agent, said agent comprising a vector having affinity for said pre-targeting vector.
33. A method as claimed in claim 32 wherein said pre-targeting vector comprises a monoclonal antibody.
34. A method as claimed in claim 31 which comprises the steps:
i) administering to said body an agent; and thereafter
ii) administering a substance capable of displacing or releasing said agent from its target.
35. A method as claimed in claim 31 wherein said agent further comprises a therapeutic compound.
36. A method as claimed in claim 35 wherein said therapeutic compound is covalently coupled or linked to the reporter through disulphide groups, and a composition comprising a reducing agent capable of reductively cleaving said disulphide groups is subsequently administered.
37. A method for in vitro investigation of targeting by an agent as defined in claim 1 wherein cells expressing a target are fixedly positioned in a flow chamber, a suspension of said agent in a carrier liquid is passed through said chamber, and binding of said agent to said cells is examined.
38. A method as claimed in claim 37 wherein the flow rate of carrier liquid is controlled to simulate shear rates encountered in vivo.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/734,730 US20050002865A1 (en) | 1996-10-28 | 2003-12-15 | Diagnostic/therapeutic agents |
Applications Claiming Priority (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9622369.8A GB9622369D0 (en) | 1996-10-28 | 1996-10-28 | Improvements in or relating to diagnostic/therapeutic agents |
| GBGB9622366.4A GB9622366D0 (en) | 1996-10-28 | 1996-10-28 | Improvements in or relating to diagnostic/therapeutic agents |
| GB9622366.4 | 1996-10-28 | ||
| GB9622369.8 | 1996-10-28 | ||
| GBGB9702195.0A GB9702195D0 (en) | 1997-02-04 | 1997-02-04 | Improvements in or relating to diagnostic/therapeutic agents |
| GB9702195.0 | 1997-02-04 | ||
| GBGB9708265.5A GB9708265D0 (en) | 1997-04-24 | 1997-04-24 | Contrast agents |
| GB9708265.5 | 1997-04-24 | ||
| US4926697P | 1997-06-06 | 1997-06-06 | |
| US4926497P | 1997-06-06 | 1997-06-06 | |
| US4926397P | 1997-06-06 | 1997-06-06 | |
| GBGB9711839.2A GB9711839D0 (en) | 1997-06-06 | 1997-06-06 | Improvements in or relating to diagnostic/therapeutic agents |
| GB9711837.6 | 1997-06-06 | ||
| GBGB9711837.6A GB9711837D0 (en) | 1997-06-06 | 1997-06-06 | Improvements in or relating to diagnostic / therapeutic agents |
| GB9711839.2 | 1997-06-06 | ||
| US08/959,206 US6331289B1 (en) | 1996-10-28 | 1997-10-28 | Targeted diagnostic/therapeutic agents having more than one different vectors |
| US09/925,715 US6680047B2 (en) | 1996-10-28 | 2001-08-10 | Diagnostic/therapeutic agents |
| US10/734,730 US20050002865A1 (en) | 1996-10-28 | 2003-12-15 | Diagnostic/therapeutic agents |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/925,715 Continuation US6680047B2 (en) | 1996-10-28 | 2001-08-10 | Diagnostic/therapeutic agents |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20050002865A1 true US20050002865A1 (en) | 2005-01-06 |
Family
ID=46149778
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US08/959,206 Expired - Fee Related US6331289B1 (en) | 1996-10-28 | 1997-10-28 | Targeted diagnostic/therapeutic agents having more than one different vectors |
| US09/925,715 Expired - Fee Related US6680047B2 (en) | 1996-10-28 | 2001-08-10 | Diagnostic/therapeutic agents |
| US10/734,730 Abandoned US20050002865A1 (en) | 1996-10-28 | 2003-12-15 | Diagnostic/therapeutic agents |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US08/959,206 Expired - Fee Related US6331289B1 (en) | 1996-10-28 | 1997-10-28 | Targeted diagnostic/therapeutic agents having more than one different vectors |
| US09/925,715 Expired - Fee Related US6680047B2 (en) | 1996-10-28 | 2001-08-10 | Diagnostic/therapeutic agents |
Country Status (1)
| Country | Link |
|---|---|
| US (3) | US6331289B1 (en) |
Cited By (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040018974A1 (en) * | 2002-03-01 | 2004-01-29 | Christophe Arbogast | Multivalent constructs for therapeutic and diagnostic applications |
| US20040210041A1 (en) * | 2002-03-01 | 2004-10-21 | Christophe Arbogast | Multivalent constructs for therapeutic and diagnostic applications |
| US20040268425A1 (en) * | 2003-03-05 | 2004-12-30 | Deliatroph Pharmaceuticals, Inc. | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US20050100963A1 (en) * | 2002-03-01 | 2005-05-12 | Dyax Corporation | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US20050147555A1 (en) * | 2002-03-01 | 2005-07-07 | Hong Fan | Methods for preparing multivalent constructs for therapeutic and diagnostic applications and methods of preparing the same |
| US20050209172A1 (en) * | 2004-03-17 | 2005-09-22 | American Pharmaceutical Partners, Inc. | Lyophilized azithromycin formulation |
| US20050250700A1 (en) * | 2002-03-01 | 2005-11-10 | Sato Aaron K | KDR and VEGF/KDR binding peptides |
| US20050260186A1 (en) * | 2003-03-05 | 2005-11-24 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20060104968A1 (en) * | 2003-03-05 | 2006-05-18 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminogly ycanases |
| US20060116336A1 (en) * | 2004-03-17 | 2006-06-01 | American Pharmaceutical Partners, Inc. | Lyophilized azithromycin formulation |
| US20070032862A1 (en) * | 2005-08-08 | 2007-02-08 | Jan Weber | Medical devices |
| US20070038176A1 (en) * | 2005-07-05 | 2007-02-15 | Jan Weber | Medical devices with machined layers for controlled communications with underlying regions |
| WO2007065016A2 (en) * | 2005-12-02 | 2007-06-07 | Au Jessie L S | Methods and compositions to improve activity and reduce toxicity of stents |
| US20070154513A1 (en) * | 2005-12-30 | 2007-07-05 | Liliana Atanasoska | Medical devices having multiple charged layers |
| US20070154466A1 (en) * | 2005-12-30 | 2007-07-05 | Jan Weber | Internal medical devices containing peroxide-converting catalysts |
| US20070224116A1 (en) * | 2006-03-27 | 2007-09-27 | Chandru Chandrasekaran | Medical devices comprising a porous metal oxide or metal material and a polymer coating for delivering therapeutic agents |
| US20070239256A1 (en) * | 2006-03-22 | 2007-10-11 | Jan Weber | Medical devices having electrical circuits with multilayer regions |
| US20070264303A1 (en) * | 2006-05-12 | 2007-11-15 | Liliana Atanasoska | Coating for medical devices comprising an inorganic or ceramic oxide and a therapeutic agent |
| US20080004691A1 (en) * | 2006-06-29 | 2008-01-03 | Boston Scientific Scimed, Inc. | Medical devices with selective coating |
| US20080086195A1 (en) * | 2006-10-05 | 2008-04-10 | Boston Scientific Scimed, Inc. | Polymer-Free Coatings For Medical Devices Formed By Plasma Electrolytic Deposition |
| US20080107607A1 (en) * | 2002-03-01 | 2008-05-08 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| US20080152594A1 (en) * | 2002-03-01 | 2008-06-26 | Philippe Bussat | Targeting vector-phospholipid conjugates |
| WO2008112297A2 (en) * | 2007-03-13 | 2008-09-18 | Yale University | Methods of treating cancer by interfering with igf-i receptor signaling |
| US20080294246A1 (en) * | 2007-05-23 | 2008-11-27 | Boston Scientific Scimed, Inc. | Endoprosthesis with Select Ceramic Morphology |
| US20090018639A1 (en) * | 2007-07-11 | 2009-01-15 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US20090018647A1 (en) * | 2007-07-11 | 2009-01-15 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US20090029077A1 (en) * | 2007-07-27 | 2009-01-29 | Boston Scientific Scimed, Inc. | Drug eluting medical devices having porous layers |
| US20090035448A1 (en) * | 2007-07-31 | 2009-02-05 | Boston Scientific Scimed, Inc. | Medical device coating by laser cladding |
| US20090099062A1 (en) * | 2007-05-31 | 2009-04-16 | Ethan Lee | Pyrvinium For The Treatment of Cancer |
| US20090104244A1 (en) * | 2007-09-21 | 2009-04-23 | Boston Scientific Scimed, Inc. | Therapeutic agent-eluting medical devices having textured polymeric surfaces |
| US20090118820A1 (en) * | 2007-11-02 | 2009-05-07 | Boston Scientific Scimed, Inc. | Deformable underlayer for stent |
| US20090118822A1 (en) * | 2007-11-02 | 2009-05-07 | Holman Thomas J | Stent with embedded material |
| US20090118818A1 (en) * | 2007-11-02 | 2009-05-07 | Boston Scientific Scimed, Inc. | Endoprosthesis with coating |
| US20090118809A1 (en) * | 2007-11-02 | 2009-05-07 | Torsten Scheuermann | Endoprosthesis with porous reservoir and non-polymer diffusion layer |
| US20090123367A1 (en) * | 2003-03-05 | 2009-05-14 | Delfmems | Soluble Glycosaminoglycanases and Methods of Preparing and Using Soluble Glycosaminoglycanases |
| US20090138077A1 (en) * | 2007-07-27 | 2009-05-28 | Boston Scientific Scimed, Inc. | Articles having ceramic coated surfaces |
| US20090324685A1 (en) * | 2008-06-26 | 2009-12-31 | Boston Scientific Scimed, Inc. | Medical device coatings containing charged materials |
| US20090324684A1 (en) * | 2008-06-25 | 2009-12-31 | Boston Scientific Scimed, Inc. | Medical devices having surface coatings |
| US20100087783A1 (en) * | 2008-10-07 | 2010-04-08 | Boston Scientific Scimed, Inc. | Medical devices for delivery of therapeutic agents to body lumens |
| US20100137978A1 (en) * | 2008-12-03 | 2010-06-03 | Boston Scientific Scimed, Inc. | Medical Implants Including Iridium Oxide |
| US20100137977A1 (en) * | 2007-08-03 | 2010-06-03 | Boston Scientific Scimed, Inc. | Coating for Medical Device Having Increased Surface Area |
| US20100228341A1 (en) * | 2009-03-04 | 2010-09-09 | Boston Scientific Scimed, Inc. | Endoprostheses |
| US20100233238A1 (en) * | 2006-03-24 | 2010-09-16 | Boston Scientific Scimed, Inc. | Medical Devices Having Nanoporous Coatings for Controlled Therapeutic Agent Delivery |
| US20100272882A1 (en) * | 2009-04-24 | 2010-10-28 | Boston Scientific Scimed, Inc. | Endoprosthese |
| US20100274352A1 (en) * | 2009-04-24 | 2010-10-28 | Boston Scientific Scrimed, Inc. | Endoprosthesis with Selective Drug Coatings |
| US20100280612A1 (en) * | 2004-12-09 | 2010-11-04 | Boston Scientific Scimed, Inc. | Medical Devices Having Vapor Deposited Nanoporous Coatings For Controlled Therapeutic Agent Delivery |
| US20100286763A1 (en) * | 1998-04-11 | 2010-11-11 | Boston Scientific Scimed, Inc. | Drug-releasing stent with ceramic-containing layer |
| US20110097275A1 (en) * | 2002-03-01 | 2011-04-28 | Bracco Suisse Sa | Kdr and vegf/kdr binding peptides and their use in diagnosis and therapy |
| US7981150B2 (en) | 2006-11-09 | 2011-07-19 | Boston Scientific Scimed, Inc. | Endoprosthesis with coatings |
| US8067054B2 (en) | 2007-04-05 | 2011-11-29 | Boston Scientific Scimed, Inc. | Stents with ceramic drug reservoir layer and methods of making and using the same |
| US8070797B2 (en) | 2007-03-01 | 2011-12-06 | Boston Scientific Scimed, Inc. | Medical device with a porous surface for delivery of a therapeutic agent |
| US8092818B2 (en) | 2006-05-17 | 2012-01-10 | Boston Scientific Scimed, Inc. | Medical devices having bioactive surfaces |
| US8216632B2 (en) | 2007-11-02 | 2012-07-10 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| EP2537532A1 (en) * | 2011-06-22 | 2012-12-26 | J. Stefan Institute | Cathepsin-binding compounds bound to a nanodevice and their diagnostic and therapeutic use |
| US8353949B2 (en) | 2006-09-14 | 2013-01-15 | Boston Scientific Scimed, Inc. | Medical devices with drug-eluting coating |
| US8431149B2 (en) | 2007-03-01 | 2013-04-30 | Boston Scientific Scimed, Inc. | Coated medical devices for abluminal drug delivery |
| US8449603B2 (en) | 2008-06-18 | 2013-05-28 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US8545865B2 (en) | 2006-03-24 | 2013-10-01 | Boston Scientific Scimed, Inc. | Medical devices having polymer brushes |
| US20140202759A1 (en) * | 2010-12-15 | 2014-07-24 | Advanced Bionics Ag | Protection for implanted gold surfaces |
| US8815275B2 (en) | 2006-06-28 | 2014-08-26 | Boston Scientific Scimed, Inc. | Coatings for medical devices comprising a therapeutic agent and a metallic material |
| US20140271901A1 (en) * | 2013-03-15 | 2014-09-18 | Mark Humayun | Management of Tractional Membranes |
| US8920491B2 (en) | 2008-04-22 | 2014-12-30 | Boston Scientific Scimed, Inc. | Medical devices having a coating of inorganic material |
| US8932346B2 (en) | 2008-04-24 | 2015-01-13 | Boston Scientific Scimed, Inc. | Medical devices having inorganic particle layers |
| US9284409B2 (en) | 2007-07-19 | 2016-03-15 | Boston Scientific Scimed, Inc. | Endoprosthesis having a non-fouling surface |
| US20180092987A1 (en) * | 2007-08-14 | 2018-04-05 | The Regents Of The University Of California | Hollow silica nanospheres and methods of making same |
| US11103593B2 (en) | 2013-10-15 | 2021-08-31 | Seagen Inc. | Pegylated drug-linkers for improved ligand-drug conjugate pharmacokinetics |
| US11229708B2 (en) | 2015-12-04 | 2022-01-25 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11730822B2 (en) | 2017-03-24 | 2023-08-22 | Seagen Inc. | Process for the preparation of glucuronide drug-linkers and intermediates thereof |
| US11793880B2 (en) | 2015-12-04 | 2023-10-24 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11844839B2 (en) | 2016-03-25 | 2023-12-19 | Seagen Inc. | Process for the preparation of pegylated drug-linkers and intermediates thereof |
| US11919836B2 (en) | 2015-09-15 | 2024-03-05 | Praxis Bioresearch, LLC | Prodrugs of fencamfamine |
Families Citing this family (94)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020150539A1 (en) * | 1989-12-22 | 2002-10-17 | Unger Evan C. | Ultrasound imaging and treatment |
| US6088613A (en) | 1989-12-22 | 2000-07-11 | Imarx Pharmaceutical Corp. | Method of magnetic resonance focused surgical and therapeutic ultrasound |
| US5205290A (en) | 1991-04-05 | 1993-04-27 | Unger Evan C | Low density microspheres and their use as contrast agents for computed tomography |
| US7083572B2 (en) * | 1993-11-30 | 2006-08-01 | Bristol-Myers Squibb Medical Imaging, Inc. | Therapeutic delivery systems |
| US6743779B1 (en) | 1994-11-29 | 2004-06-01 | Imarx Pharmaceutical Corp. | Methods for delivering compounds into a cell |
| US6033645A (en) * | 1996-06-19 | 2000-03-07 | Unger; Evan C. | Methods for diagnostic imaging by regulating the administration rate of a contrast agent |
| US6521211B1 (en) * | 1995-06-07 | 2003-02-18 | Bristol-Myers Squibb Medical Imaging, Inc. | Methods of imaging and treatment with targeted compositions |
| US20050019266A1 (en) * | 1997-05-06 | 2005-01-27 | Unger Evan C. | Novel targeted compositions for diagnostic and therapeutic use |
| US6416740B1 (en) * | 1997-05-13 | 2002-07-09 | Bristol-Myers Squibb Medical Imaging, Inc. | Acoustically active drug delivery systems |
| US6548047B1 (en) | 1997-09-15 | 2003-04-15 | Bristol-Myers Squibb Medical Imaging, Inc. | Thermal preactivation of gaseous precursor filled compositions |
| US20010003580A1 (en) | 1998-01-14 | 2001-06-14 | Poh K. Hui | Preparation of a lipid blend and a phospholipid suspension containing the lipid blend |
| ATE276745T1 (en) * | 1998-04-09 | 2004-10-15 | Amersham Health As | USE OF PARTICLE CONTRAST AGENTS IN DIAGNOSTIC IMAGING FOR THE INVESTIGATION OF PHYSIOLOGICAL PARAMETERS |
| US9616150B2 (en) * | 1999-10-29 | 2017-04-11 | Children's Hospital Los Angeles | Bone hemostasis method and materials |
| US6570196B1 (en) * | 2000-03-22 | 2003-05-27 | Max-Plank-Gesellschaft Zur Forderung Der Wissenschaften | Lipid vesicles or lipid bilayers on chips |
| WO2002062810A2 (en) * | 2000-11-29 | 2002-08-15 | Bracco International B.V. | Linkable sialyl lewis x analogs |
| WO2002062957A2 (en) * | 2001-02-07 | 2002-08-15 | Invitrogen Corporation | Ter sites and ter binding proteins |
| US7198945B2 (en) | 2001-08-09 | 2007-04-03 | Teruyuki Nagamune | Cell having modified cell membrane |
| US20030044354A1 (en) * | 2001-08-16 | 2003-03-06 | Carpenter Alan P. | Gas microsphere liposome composites for ultrasound imaging and ultrasound stimulated drug release |
| US7087212B2 (en) | 2001-08-17 | 2006-08-08 | Mallinckrodt, Inc | Multicomponent assemblies having enhanced binding properties for diagnosis and therapy |
| ES2290333T3 (en) * | 2001-11-13 | 2008-02-16 | Celator Pharmaceuticals, Inc. | COMPOSITIONS OF LIPID VEHICLES AND METHODS FOR THE IMPROVED RETENTION OF PHARMACOS. |
| EP1489971A2 (en) * | 2002-04-03 | 2004-12-29 | Jackie R. See | Methods for ultrasonic imaging and treating diseased tissues |
| US20030232016A1 (en) * | 2002-04-17 | 2003-12-18 | Russell Heinrich | Nerve identification and sparing method |
| AU2003257109A1 (en) * | 2002-08-05 | 2004-02-23 | Invitrogen Corporation | Compositions and methods for molecular biology |
| NO20024755D0 (en) * | 2002-10-03 | 2002-10-03 | Amersham Health As | Method |
| KR101025143B1 (en) * | 2002-12-31 | 2011-04-01 | 넥타르 테라퓨틱스 | Hydrolytically stable maleimide-terminated polymers |
| US7432331B2 (en) | 2002-12-31 | 2008-10-07 | Nektar Therapeutics Al, Corporation | Hydrolytically stable maleimide-terminated polymers |
| US7500953B2 (en) * | 2003-01-25 | 2009-03-10 | Seno Medical Instruments, Inc. | High contrast optoacoustic imaging using nanoparticles |
| US20070128117A1 (en) * | 2003-02-04 | 2007-06-07 | Bracco International B.V. | Ultrasound contrast agents and process for the preparation thereof |
| DE602004029010D1 (en) * | 2003-02-04 | 2010-10-21 | Bracco Suisse Sa | ULTRASONIC CONTRASTING AGENT AND METHOD OF CREATION |
| ES2695401T3 (en) * | 2003-02-12 | 2019-01-04 | Syncera Inc | Alloxide compositions of random and non-random alkylene oxide polymers |
| ES2396368T3 (en) | 2003-03-03 | 2013-02-21 | Dyax Corporation | Peptides that specifically bind to the HGF receptor (CMET) and uses thereof |
| NO20033115D0 (en) * | 2003-07-08 | 2003-07-08 | Amersham Health As | Peptide-based compounds |
| US20050187278A1 (en) * | 2003-08-28 | 2005-08-25 | Pharmacia Corporation | Treatment or prevention of vascular disorders with Cox-2 inhibitors in combination with cyclic AMP-specific phosphodiesterase inhibitors |
| WO2005023767A2 (en) | 2003-09-10 | 2005-03-17 | Brentwood Equities Ltd. | Diastereomers of 4-aryloxy-3-hydroxypiperidines |
| US7314466B2 (en) * | 2003-10-02 | 2008-01-01 | Lary G Banning | Minimally invasive vascular surgery |
| IL158658A0 (en) * | 2003-10-29 | 2004-05-12 | Moshe Schwarzberg | Prokinetic drugs assistance to small intestine imaging |
| WO2005063305A1 (en) * | 2003-12-22 | 2005-07-14 | Bracco Research Sa | Gas-filled microvesicle assembly for contrast imaging |
| CN1897978B (en) * | 2003-12-22 | 2011-11-23 | 博莱科瑞士股份有限公司 | Assembly of gas-filled microvesicle with active component for contrast imaging |
| DE602005021057D1 (en) | 2004-01-20 | 2010-06-17 | Toronto E | HIGH FREQUENCY ULTRASONIC PRESENTATION WITH CONTRAST |
| US7555456B2 (en) * | 2004-06-08 | 2009-06-30 | Rosenthal Collins Group, Llc | Method and system for providing electronic information for multi-market electronic trading |
| FR2873703B1 (en) * | 2004-07-30 | 2006-12-08 | Flamel Technologies Sa | BRANCHED POLYAMINOACIDES FUNCTIONALIZED BY HYDROPHOBIC GROUPS AND THEIR PARTICULARLY THERAPEUTIC APPLICATIONS |
| EP1877097B1 (en) * | 2004-08-11 | 2012-06-20 | Arqule, Inc. | Aminoacid conjugates of beta-lapachone for tumor targeting |
| EP1784228B1 (en) | 2004-08-18 | 2016-10-05 | Bracco Suisse SA | Gas-filled microvesicles composition for contrast imaging |
| US7888346B2 (en) * | 2004-09-24 | 2011-02-15 | Universtiy of Maryland, Baltimore | Method of treating organophosphorous poisoning |
| US9132135B2 (en) * | 2004-09-24 | 2015-09-15 | University Of Maryland, Baltimore | Method of treating organophosphorous poisoning |
| CN101438252A (en) * | 2004-10-07 | 2009-05-20 | 爱莫里大学 | Multifunctional nanoparticle conjugates and their use |
| US8034222B2 (en) * | 2004-10-26 | 2011-10-11 | The Regents Of The University Of California | Conducting polymer nanowire sensors |
| WO2006063016A2 (en) | 2004-12-09 | 2006-06-15 | Rosenthal Collins Group, Llc | Method and system for providing configurable features for graphical user interfaces for electronic trading |
| US20120269886A1 (en) | 2004-12-22 | 2012-10-25 | Nitto Denko Corporation | Therapeutic agent for pulmonary fibrosis |
| LT2727583T (en) | 2004-12-22 | 2021-12-27 | Nitto Denko Corporation | Drug carrier and drug carrier kit for inhibiting fibrosis |
| US20070059775A1 (en) * | 2005-03-29 | 2007-03-15 | The Trustees Of Columbia University In The City Of New York | Synthesis and conjugation of iron oxide nanoparticles to antibodies for targeting specific cells using fluorescence and MR imaging techniques |
| MX2007012486A (en) * | 2005-04-08 | 2007-12-13 | Mycosol Inc | Stilbazium research assays. |
| US7801801B2 (en) | 2005-05-04 | 2010-09-21 | Rosenthal Collins Group, Llc | Method and system for providing automatic execution of black box strategies for electonic trading |
| US7815575B2 (en) * | 2005-05-09 | 2010-10-19 | Salutron, Inc. | Ultrasonic monitor with a biocompatible oil based transmission medium |
| EP1896500A4 (en) * | 2005-06-28 | 2010-08-11 | Yeda Res & Dev | Gliomedin, fragments thereof and methods of using same |
| WO2007056553A2 (en) | 2005-11-13 | 2007-05-18 | Rosenthal Collins Group, Llc | Method and system for electronic trading via a yield curve |
| US9572886B2 (en) | 2005-12-22 | 2017-02-21 | Nitto Denko Corporation | Agent for treating myelofibrosis |
| JP5872751B2 (en) * | 2006-02-06 | 2016-03-01 | バーナム インスティテュート フォー メディカル リサーチ | Methods and compositions for tumor and injury targeting |
| WO2007097318A1 (en) * | 2006-02-23 | 2007-08-30 | Adeka Corporation | Diagnostic marker |
| US8367065B2 (en) * | 2006-09-15 | 2013-02-05 | Enzon Pharmaceuticals, Inc. | Targeted polymeric prodrugs containing multifunctional linkers |
| JP2010503708A (en) * | 2006-09-15 | 2010-02-04 | エンゾン ファーマスーティカルズ インコーポレイテッド | Targeted polymer prodrugs containing multifunctional linkers |
| TWI407971B (en) | 2007-03-30 | 2013-09-11 | Nitto Denko Corp | Cancer cells and tumor-related fibroblasts |
| US20100150924A1 (en) * | 2007-05-22 | 2010-06-17 | Elior Peles | Regulation of myelination by nectin-like (necl) molecules |
| DE102007041834A1 (en) * | 2007-09-03 | 2009-03-05 | Siemens Ag | Medicines for the treatment of a carcinoma |
| JP2010539245A (en) | 2007-09-14 | 2010-12-16 | 日東電工株式会社 | Drug carrier |
| US20090170770A1 (en) * | 2007-11-06 | 2009-07-02 | Ali Hafezi-Moghadam | Methods and compositions for treating conditions associated with angiogenesis using a vascular adhesion protein-1 (vap 1) inhibitor |
| EP2087930A1 (en) * | 2008-02-05 | 2009-08-12 | Evonik Degussa GmbH | Method for the absorption of volatile material in a fluid absorption agent |
| EP2088389B1 (en) * | 2008-02-05 | 2017-05-10 | Evonik Degussa GmbH | Absorption cooling machine |
| WO2010062854A2 (en) * | 2008-11-26 | 2010-06-03 | Arizona Board Of Regents, A Body Corporate Of The State Of Arizona, Acting For And On Behalf Of Arizona State University | Methods and compositions for using bleomycin-derivatized microbubbles |
| ES2382236T3 (en) * | 2009-06-05 | 2012-06-06 | Evonik Degussa Gmbh | Procedure, absorption medium and device for CO2 absorption from gas mixtures |
| US10058837B2 (en) | 2009-08-28 | 2018-08-28 | The Trustees Of Columbia University In The City Of New York | Systems, methods, and devices for production of gas-filled microbubbles |
| WO2012030675A1 (en) * | 2010-08-28 | 2012-03-08 | The Trustees Of Columbia University In The City Of New York | Systems, methods, and devices for plasmid gene transfection using polymer-modified microbubbles |
| DE102011077377A1 (en) | 2010-11-12 | 2012-05-16 | Evonik Degussa Gmbh | Process for the absorption of acid gases from gas mixtures |
| WO2012136813A2 (en) | 2011-04-07 | 2012-10-11 | Universitetet I Oslo | Agents for medical radar diagnosis |
| WO2013072147A1 (en) | 2011-11-14 | 2013-05-23 | Evonik Degussa Gmbh | Method and device for the separation of acidic gases from a gas mixture |
| DE102012200907A1 (en) | 2012-01-23 | 2013-07-25 | Evonik Industries Ag | Method and absorption medium for absorbing CO2 from a gas mixture |
| US20150132227A1 (en) * | 2012-04-18 | 2015-05-14 | University Of Utah Research Foundation | Novel echogenic contrast agents |
| DE102012207509A1 (en) | 2012-05-07 | 2013-11-07 | Evonik Degussa Gmbh | Method for absorbing CO2 from a gas mixture |
| WO2013188755A2 (en) * | 2012-06-14 | 2013-12-19 | The Board Of Trustees Of The University Of Arkansas | Compositions and methods for non-invasive detection and treatment of hypoxic cells in vivo |
| CN102813942A (en) * | 2012-08-15 | 2012-12-12 | 钟志容 | Lipid ultrasound microbubble mediated adeno-associated virus gene transfection preparation and preparation technology thereof |
| US10265424B2 (en) | 2014-03-04 | 2019-04-23 | Trust-Biosonics, Inc. | Molecular imaging contrast agents and uses thereof |
| US11291931B2 (en) | 2014-12-15 | 2022-04-05 | Akadeum Life Sciences, Inc. | Method and system for buoyant separation |
| DE102015212749A1 (en) | 2015-07-08 | 2017-01-12 | Evonik Degussa Gmbh | Method for dehumidifying moist gas mixtures |
| TWI616210B (en) | 2016-01-07 | 2018-03-01 | 國立臺灣科技大學 | Modified microbubble and method of making the same |
| DE102016210478A1 (en) | 2016-06-14 | 2017-12-14 | Evonik Degussa Gmbh | Method for dehumidifying moist gas mixtures |
| EP3257568B1 (en) | 2016-06-14 | 2019-09-18 | Evonik Degussa GmbH | Method for the removal of moisture from moist gas mixtures by use of ionic liquids |
| DE102016210483A1 (en) | 2016-06-14 | 2017-12-14 | Evonik Degussa Gmbh | Process and absorbent for dehumidifying moist gas mixtures |
| EP3257843A1 (en) | 2016-06-14 | 2017-12-20 | Evonik Degussa GmbH | Method of preparing a high purity imidazolium salt |
| DE102016210481B3 (en) | 2016-06-14 | 2017-06-08 | Evonik Degussa Gmbh | Process for purifying an ionic liquid |
| DE102016210484A1 (en) | 2016-06-14 | 2017-12-14 | Evonik Degussa Gmbh | Method for dehumidifying moist gas mixtures |
| EP3820588A4 (en) | 2018-07-09 | 2022-04-06 | Akadeum Life Sciences, Inc. | System and method for buoyant particle processing |
| US11292803B2 (en) | 2018-08-21 | 2022-04-05 | The Board Of Trustees Of The University Of Illinois | Bioreducible N-oxide-based probes for imaging of hypoxia |
| US11857551B1 (en) | 2020-07-10 | 2024-01-02 | Ting Therapeutics Llc | Methods for the prevention and treatment of hearing loss |
| US11819842B2 (en) | 2021-08-26 | 2023-11-21 | Akadeum Life Sciences, Inc. | Method and system for buoyant separation |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4927916A (en) * | 1984-04-23 | 1990-05-22 | The General Hospital Corporation | Method of producing fibrin-specific monoclonal antibodies lacking fibrinogen-cross-reactivity using fibrin-specific peptides |
| US5013556A (en) * | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5154924A (en) * | 1989-09-07 | 1992-10-13 | Alkermes, Inc. | Transferrin receptor specific antibody-neuropharmaceutical agent conjugates |
| US5356633A (en) * | 1989-10-20 | 1994-10-18 | Liposome Technology, Inc. | Method of treatment of inflamed tissues |
| US5362478A (en) * | 1993-03-26 | 1994-11-08 | Vivorx Pharmaceuticals, Inc. | Magnetic resonance imaging with fluorocarbons encapsulated in a cross-linked polymeric shell |
| US5498421A (en) * | 1993-02-22 | 1996-03-12 | Vivorx Pharmaceuticals, Inc. | Composition useful for in vivo delivery of biologics and methods employing same |
| US5846517A (en) * | 1996-09-11 | 1998-12-08 | Imarx Pharmaceutical Corp. | Methods for diagnostic imaging using a renal contrast agent and a vasodilator |
| US5849727A (en) * | 1996-06-28 | 1998-12-15 | Board Of Regents Of The University Of Nebraska | Compositions and methods for altering the biodistribution of biological agents |
| US5910300A (en) * | 1995-11-01 | 1999-06-08 | Bracco Research S.A. | Amphiphilic linkers for coupling administrable diagnostically or physiologically active agents and bioselective targeting compounds |
| US6245318B1 (en) * | 1997-05-27 | 2001-06-12 | Mallinckrodt Inc. | Selectively binding ultrasound contrast agents |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5198424A (en) | 1989-03-08 | 1993-03-30 | Board Of Regents Of The University Of Oklahoma | Functionally active selectin-derived peptides |
| US5656211A (en) | 1989-12-22 | 1997-08-12 | Imarx Pharmaceutical Corp. | Apparatus and method for making gas-filled vesicles of optimal size |
| US5542935A (en) | 1989-12-22 | 1996-08-06 | Imarx Pharmaceutical Corp. | Therapeutic delivery systems related applications |
| US5580575A (en) | 1989-12-22 | 1996-12-03 | Imarx Pharmaceutical Corp. | Therapeutic drug delivery systems |
| US5733572A (en) | 1989-12-22 | 1998-03-31 | Imarx Pharmaceutical Corp. | Gas and gaseous precursor filled microspheres as topical and subcutaneous delivery vehicles |
| IN172208B (en) | 1990-04-02 | 1993-05-01 | Sint Sa | |
| US5632986A (en) | 1991-05-09 | 1997-05-27 | The University Of Washington | Phospholipid-targeted thrombolytic agents |
| WO1993020802A1 (en) | 1992-04-09 | 1993-10-28 | Northwestern University | Acoustically reflective liposomes and methods to make and use the same |
| US6217869B1 (en) * | 1992-06-09 | 2001-04-17 | Neorx Corporation | Pretargeting methods and compounds |
| DE4232755A1 (en) | 1992-09-26 | 1994-03-31 | Schering Ag | Microparticle preparations made from biodegradable copolymers |
| US5650156A (en) | 1993-02-22 | 1997-07-22 | Vivorx Pharmaceuticals, Inc. | Methods for in vivo delivery of nutriceuticals and compositions useful therefor |
| US5665383A (en) | 1993-02-22 | 1997-09-09 | Vivorx Pharmaceuticals, Inc. | Methods for the preparation of immunostimulating agents for in vivo delivery |
| US5534241A (en) | 1993-07-23 | 1996-07-09 | Torchilin; Vladimir P. | Amphipathic polychelating compounds and methods of use |
| US5565215A (en) | 1993-07-23 | 1996-10-15 | Massachusettes Institute Of Technology | Biodegradable injectable particles for imaging |
| CA2167920A1 (en) | 1993-07-23 | 1995-02-02 | Abraham J. Domb | Nonoparticles and microparticles of non-linear hydrophilic-hydrophobic multiblock copolymers |
| AU7655194A (en) | 1993-09-09 | 1995-03-27 | Schering Aktiengesellschaft | Active principles and gas containing microparticles |
| US5716594A (en) | 1994-06-06 | 1998-02-10 | The Jmde Trust | Biotin compounds for targetting tumors and sites of infection |
| EP0727225A3 (en) | 1995-02-14 | 1997-01-15 | Sonus Pharma Inc | Compositions and methods for directed ultrasound imaging |
| WO1996039149A1 (en) | 1995-06-06 | 1996-12-12 | Merck & Co., Inc. | Anhydrous alendronate monosodium salt formulations |
| US5997898A (en) | 1995-06-06 | 1999-12-07 | Imarx Pharmaceutical Corp. | Stabilized compositions of fluorinated amphiphiles for methods of therapeutic delivery |
| WO1996040277A2 (en) | 1995-06-07 | 1996-12-19 | Brown University Research Foundation | Spray dried polymeric microparticles containing imaging agents |
| ATE265863T1 (en) | 1995-06-07 | 2004-05-15 | Imarx Pharmaceutical Corp | NEW TARGETED AGENTS FOR DIAGNOSTIC AND THERAPEUTIC USE |
| US5690907A (en) | 1995-06-08 | 1997-11-25 | The Jewish Hospital Of St. Louis | Avidin-biotin conjugated emulsions as a site specific binding system |
| US5780010A (en) | 1995-06-08 | 1998-07-14 | Barnes-Jewish Hospital | Method of MRI using avidin-biotin conjugated emulsions as a site specific binding system |
| DE19549240A1 (en) | 1995-12-21 | 1997-07-10 | Schering Ag | Portable device for the simulation of ultrasound examinations |
| WO1997033474A1 (en) | 1996-03-12 | 1997-09-18 | Board Of Regents Of The University Of Nebraska | Targeted site specific drug delivery compositions and method of use |
| JP2000510119A (en) | 1996-05-03 | 2000-08-08 | イムノメディクス,インコーポレイテッド | Targeted combination immunotherapy for cancer |
| DE19626530A1 (en) | 1996-07-02 | 1998-01-15 | Byk Gulden Lomberg Chem Fab | Aqueous magnetic resonance contrast agent compositions |
| AU4047497A (en) | 1996-07-31 | 1998-02-20 | Immunomedics Inc. | Improved detection and therapy of lesions with biotin-chelate conjugates |
| TW520297B (en) | 1996-10-11 | 2003-02-11 | Sequus Pharm Inc | Fusogenic liposome composition and method |
| EP0941120A4 (en) | 1996-11-05 | 2004-08-18 | Bristol Myers Squibb Co | Branched peptide linkers |
| DE19648664A1 (en) | 1996-11-14 | 1998-05-28 | Schering Ag | Microparticles containing active ingredients, compositions containing them, their use for the ultrasound-controlled release of active ingredients and processes for their production |
| US6143276A (en) | 1997-03-21 | 2000-11-07 | Imarx Pharmaceutical Corp. | Methods for delivering bioactive agents to regions of elevated temperatures |
-
1997
- 1997-10-28 US US08/959,206 patent/US6331289B1/en not_active Expired - Fee Related
-
2001
- 2001-08-10 US US09/925,715 patent/US6680047B2/en not_active Expired - Fee Related
-
2003
- 2003-12-15 US US10/734,730 patent/US20050002865A1/en not_active Abandoned
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4927916A (en) * | 1984-04-23 | 1990-05-22 | The General Hospital Corporation | Method of producing fibrin-specific monoclonal antibodies lacking fibrinogen-cross-reactivity using fibrin-specific peptides |
| US5154924A (en) * | 1989-09-07 | 1992-10-13 | Alkermes, Inc. | Transferrin receptor specific antibody-neuropharmaceutical agent conjugates |
| US5013556A (en) * | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5356633A (en) * | 1989-10-20 | 1994-10-18 | Liposome Technology, Inc. | Method of treatment of inflamed tissues |
| US5498421A (en) * | 1993-02-22 | 1996-03-12 | Vivorx Pharmaceuticals, Inc. | Composition useful for in vivo delivery of biologics and methods employing same |
| US5362478A (en) * | 1993-03-26 | 1994-11-08 | Vivorx Pharmaceuticals, Inc. | Magnetic resonance imaging with fluorocarbons encapsulated in a cross-linked polymeric shell |
| US5505932A (en) * | 1993-03-26 | 1996-04-09 | Vivorx Pharmaceuticals, Inc. | Method for the preparation of fluorocarbon-containing polymeric shells for medical imaging |
| US5910300A (en) * | 1995-11-01 | 1999-06-08 | Bracco Research S.A. | Amphiphilic linkers for coupling administrable diagnostically or physiologically active agents and bioselective targeting compounds |
| US5849727A (en) * | 1996-06-28 | 1998-12-15 | Board Of Regents Of The University Of Nebraska | Compositions and methods for altering the biodistribution of biological agents |
| US5846517A (en) * | 1996-09-11 | 1998-12-08 | Imarx Pharmaceutical Corp. | Methods for diagnostic imaging using a renal contrast agent and a vasodilator |
| US6245318B1 (en) * | 1997-05-27 | 2001-06-12 | Mallinckrodt Inc. | Selectively binding ultrasound contrast agents |
Cited By (144)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100286763A1 (en) * | 1998-04-11 | 2010-11-11 | Boston Scientific Scimed, Inc. | Drug-releasing stent with ceramic-containing layer |
| US8066763B2 (en) | 1998-04-11 | 2011-11-29 | Boston Scientific Scimed, Inc. | Drug-releasing stent with ceramic-containing layer |
| US20100003195A1 (en) * | 2002-03-01 | 2010-01-07 | Sato Aaron K | Kdr and vegf/kdr binding peptides and their use in diagnosis and therapy |
| US9629934B2 (en) | 2002-03-01 | 2017-04-25 | Dyax Corp. | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US20050100963A1 (en) * | 2002-03-01 | 2005-05-12 | Dyax Corporation | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US20050147555A1 (en) * | 2002-03-01 | 2005-07-07 | Hong Fan | Methods for preparing multivalent constructs for therapeutic and diagnostic applications and methods of preparing the same |
| US8632753B2 (en) | 2002-03-01 | 2014-01-21 | Bracco Suisse Sa | Multivalent constructs for therapeutic and diagnostic applications |
| US20050250700A1 (en) * | 2002-03-01 | 2005-11-10 | Sato Aaron K | KDR and VEGF/KDR binding peptides |
| US20040210041A1 (en) * | 2002-03-01 | 2004-10-21 | Christophe Arbogast | Multivalent constructs for therapeutic and diagnostic applications |
| US8623822B2 (en) | 2002-03-01 | 2014-01-07 | Bracco Suisse Sa | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US8642010B2 (en) | 2002-03-01 | 2014-02-04 | Dyax Corp. | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US7985402B2 (en) | 2002-03-01 | 2011-07-26 | Bracco Suisse Sa | Targeting vector-phospholipid conjugates |
| US8663603B2 (en) | 2002-03-01 | 2014-03-04 | Bracco Suisse Sa | Multivalent constructs for therapeutic and diagnostic applications |
| US9381258B2 (en) | 2002-03-01 | 2016-07-05 | Bracco Suisse S.A. | Targeting vector-phospholipid conjugates |
| US7211240B2 (en) | 2002-03-01 | 2007-05-01 | Bracco International B.V. | Multivalent constructs for therapeutic and diagnostic applications |
| US20110097275A1 (en) * | 2002-03-01 | 2011-04-28 | Bracco Suisse Sa | Kdr and vegf/kdr binding peptides and their use in diagnosis and therapy |
| US20040018974A1 (en) * | 2002-03-01 | 2004-01-29 | Christophe Arbogast | Multivalent constructs for therapeutic and diagnostic applications |
| US20110070165A1 (en) * | 2002-03-01 | 2011-03-24 | Christophe Arbogast | Multivalent constructs for therapeutic and diagnostic applications |
| US7261876B2 (en) | 2002-03-01 | 2007-08-28 | Bracco International Bv | Multivalent constructs for therapeutic and diagnostic applications |
| US7910088B2 (en) | 2002-03-01 | 2011-03-22 | Bracco Suisse Sa | Multivalent constructs for therapeutic and diagnostic applications |
| US7854919B2 (en) | 2002-03-01 | 2010-12-21 | Bracco, Suisse SA | Multivalent constructs for therapeutic and diagnostic applications |
| US20100233090A1 (en) * | 2002-03-01 | 2010-09-16 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| US7794693B2 (en) | 2002-03-01 | 2010-09-14 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| US9056138B2 (en) | 2002-03-01 | 2015-06-16 | Bracco Suisse Sa | Multivalent constructs for therapeutic and diagnostic applications |
| US20080107607A1 (en) * | 2002-03-01 | 2008-05-08 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| US20080152594A1 (en) * | 2002-03-01 | 2008-06-26 | Philippe Bussat | Targeting vector-phospholipid conjugates |
| US7666979B2 (en) | 2002-03-01 | 2010-02-23 | Bracco International B.V. | Methods for preparing multivalent constructs for therapeutic and diagnostic applications and methods of preparing the same |
| US20050027105A9 (en) * | 2002-03-01 | 2005-02-03 | Christophe Arbogast | Multivalent constructs for therapeutic and diagnostic applications |
| US9295737B2 (en) | 2002-03-01 | 2016-03-29 | Bracco Suisse Sa | Targeting vector-phospholipid conjugates |
| US8551450B2 (en) | 2002-03-01 | 2013-10-08 | Philippe Bussat | Targeting vector-phospholipid conjugates |
| US9408926B2 (en) | 2002-03-01 | 2016-08-09 | Bracco Suisse S.A. | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US9446155B2 (en) | 2002-03-01 | 2016-09-20 | Bracco Suisse Sa | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US20110008309A1 (en) * | 2003-03-05 | 2011-01-13 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20060104968A1 (en) * | 2003-03-05 | 2006-05-18 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminogly ycanases |
| US8105586B2 (en) | 2003-03-05 | 2012-01-31 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20040268425A1 (en) * | 2003-03-05 | 2004-12-30 | Deliatroph Pharmaceuticals, Inc. | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US7871607B2 (en) | 2003-03-05 | 2011-01-18 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US9562223B2 (en) | 2003-03-05 | 2017-02-07 | Halozyme, Inc. | Methods for reducing intraocular pressure by administering a soluble hyaluronidase glycoprotein (sHASEGP) |
| US8450470B2 (en) | 2003-03-05 | 2013-05-28 | Halozyme, Inc. | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US8431124B2 (en) | 2003-03-05 | 2013-04-30 | Halozyme, Inc. | Methods for treating a disease characterized by an excess of hyaluronan by administering a soluble hyaluronidase glycoprotein (sHASEGP) |
| US20090123367A1 (en) * | 2003-03-05 | 2009-05-14 | Delfmems | Soluble Glycosaminoglycanases and Methods of Preparing and Using Soluble Glycosaminoglycanases |
| US20090253175A1 (en) * | 2003-03-05 | 2009-10-08 | Bookbinder Louis H | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US20090181032A1 (en) * | 2003-03-05 | 2009-07-16 | Bookbinder Louis H | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US20090181013A1 (en) * | 2003-03-05 | 2009-07-16 | Bookbinder Louis H | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US7767429B2 (en) | 2003-03-05 | 2010-08-03 | Halozyme, Inc. | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US20090214505A1 (en) * | 2003-03-05 | 2009-08-27 | Bookbinder Louis H | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US8202517B2 (en) | 2003-03-05 | 2012-06-19 | Halozyme, Inc. | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US20050260186A1 (en) * | 2003-03-05 | 2005-11-24 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US8431380B2 (en) | 2003-03-05 | 2013-04-30 | Halozyme, Inc. | Soluble hyaluronidase glycoprotein (sHASEGP), process for preparing the same, uses and pharmaceutical compositions comprising thereof |
| US8580252B2 (en) | 2004-03-05 | 2013-11-12 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US10016491B2 (en) | 2004-03-05 | 2018-07-10 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US8257699B2 (en) | 2004-03-05 | 2012-09-04 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US9211315B2 (en) | 2004-03-05 | 2015-12-15 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20060116336A1 (en) * | 2004-03-17 | 2006-06-01 | American Pharmaceutical Partners, Inc. | Lyophilized azithromycin formulation |
| US20050209172A1 (en) * | 2004-03-17 | 2005-09-22 | American Pharmaceutical Partners, Inc. | Lyophilized azithromycin formulation |
| US20100280612A1 (en) * | 2004-12-09 | 2010-11-04 | Boston Scientific Scimed, Inc. | Medical Devices Having Vapor Deposited Nanoporous Coatings For Controlled Therapeutic Agent Delivery |
| WO2006091871A1 (en) | 2005-02-23 | 2006-08-31 | Halozyme Therapeutics, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20100196423A1 (en) * | 2005-02-23 | 2010-08-05 | Bookbinder Louis H | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US10588983B2 (en) | 2005-02-23 | 2020-03-17 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| EP3943501A1 (en) | 2005-02-23 | 2022-01-26 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US7829081B2 (en) | 2005-02-23 | 2010-11-09 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| EP3045472A1 (en) | 2005-02-23 | 2016-07-20 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US7846431B2 (en) | 2005-02-23 | 2010-12-07 | Halozyme, Inc. | Soluble glycosaminoglycanases and methods of preparing and using soluble glycosaminoglycanases |
| US20070038176A1 (en) * | 2005-07-05 | 2007-02-15 | Jan Weber | Medical devices with machined layers for controlled communications with underlying regions |
| US7778684B2 (en) | 2005-08-08 | 2010-08-17 | Boston Scientific Scimed, Inc. | MRI resonator system with stent implant |
| US20070032862A1 (en) * | 2005-08-08 | 2007-02-08 | Jan Weber | Medical devices |
| WO2007065016A2 (en) * | 2005-12-02 | 2007-06-07 | Au Jessie L S | Methods and compositions to improve activity and reduce toxicity of stents |
| WO2007065016A3 (en) * | 2005-12-02 | 2009-01-08 | Jessie L S Au | Methods and compositions to improve activity and reduce toxicity of stents |
| US8834912B2 (en) | 2005-12-30 | 2014-09-16 | Boston Scientific Scimed, Inc. | Medical devices having multiple charged layers |
| US20070154466A1 (en) * | 2005-12-30 | 2007-07-05 | Jan Weber | Internal medical devices containing peroxide-converting catalysts |
| US20070154513A1 (en) * | 2005-12-30 | 2007-07-05 | Liliana Atanasoska | Medical devices having multiple charged layers |
| US20070239256A1 (en) * | 2006-03-22 | 2007-10-11 | Jan Weber | Medical devices having electrical circuits with multilayer regions |
| US20100233238A1 (en) * | 2006-03-24 | 2010-09-16 | Boston Scientific Scimed, Inc. | Medical Devices Having Nanoporous Coatings for Controlled Therapeutic Agent Delivery |
| US8574615B2 (en) | 2006-03-24 | 2013-11-05 | Boston Scientific Scimed, Inc. | Medical devices having nanoporous coatings for controlled therapeutic agent delivery |
| US8545865B2 (en) | 2006-03-24 | 2013-10-01 | Boston Scientific Scimed, Inc. | Medical devices having polymer brushes |
| US20070224116A1 (en) * | 2006-03-27 | 2007-09-27 | Chandru Chandrasekaran | Medical devices comprising a porous metal oxide or metal material and a polymer coating for delivering therapeutic agents |
| US8187620B2 (en) | 2006-03-27 | 2012-05-29 | Boston Scientific Scimed, Inc. | Medical devices comprising a porous metal oxide or metal material and a polymer coating for delivering therapeutic agents |
| US20070264303A1 (en) * | 2006-05-12 | 2007-11-15 | Liliana Atanasoska | Coating for medical devices comprising an inorganic or ceramic oxide and a therapeutic agent |
| US20110189377A1 (en) * | 2006-05-12 | 2011-08-04 | Boston Scientific Scimed, Inc. | Coating for Medical Devices Comprising An Inorganic or Ceramic Oxide and a Therapeutic Agent |
| US8092818B2 (en) | 2006-05-17 | 2012-01-10 | Boston Scientific Scimed, Inc. | Medical devices having bioactive surfaces |
| US8815275B2 (en) | 2006-06-28 | 2014-08-26 | Boston Scientific Scimed, Inc. | Coatings for medical devices comprising a therapeutic agent and a metallic material |
| US20080004691A1 (en) * | 2006-06-29 | 2008-01-03 | Boston Scientific Scimed, Inc. | Medical devices with selective coating |
| US8771343B2 (en) | 2006-06-29 | 2014-07-08 | Boston Scientific Scimed, Inc. | Medical devices with selective titanium oxide coatings |
| US8353949B2 (en) | 2006-09-14 | 2013-01-15 | Boston Scientific Scimed, Inc. | Medical devices with drug-eluting coating |
| US20080086195A1 (en) * | 2006-10-05 | 2008-04-10 | Boston Scientific Scimed, Inc. | Polymer-Free Coatings For Medical Devices Formed By Plasma Electrolytic Deposition |
| US7981150B2 (en) | 2006-11-09 | 2011-07-19 | Boston Scientific Scimed, Inc. | Endoprosthesis with coatings |
| US8431149B2 (en) | 2007-03-01 | 2013-04-30 | Boston Scientific Scimed, Inc. | Coated medical devices for abluminal drug delivery |
| US8070797B2 (en) | 2007-03-01 | 2011-12-06 | Boston Scientific Scimed, Inc. | Medical device with a porous surface for delivery of a therapeutic agent |
| WO2008112297A3 (en) * | 2007-03-13 | 2008-10-30 | Univ Yale | Methods of treating cancer by interfering with igf-i receptor signaling |
| WO2008112297A2 (en) * | 2007-03-13 | 2008-09-18 | Yale University | Methods of treating cancer by interfering with igf-i receptor signaling |
| US8067054B2 (en) | 2007-04-05 | 2011-11-29 | Boston Scientific Scimed, Inc. | Stents with ceramic drug reservoir layer and methods of making and using the same |
| US7976915B2 (en) | 2007-05-23 | 2011-07-12 | Boston Scientific Scimed, Inc. | Endoprosthesis with select ceramic morphology |
| US20080294246A1 (en) * | 2007-05-23 | 2008-11-27 | Boston Scientific Scimed, Inc. | Endoprosthesis with Select Ceramic Morphology |
| US20090099062A1 (en) * | 2007-05-31 | 2009-04-16 | Ethan Lee | Pyrvinium For The Treatment of Cancer |
| US8002823B2 (en) | 2007-07-11 | 2011-08-23 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US7942926B2 (en) | 2007-07-11 | 2011-05-17 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US20090018647A1 (en) * | 2007-07-11 | 2009-01-15 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US20090018639A1 (en) * | 2007-07-11 | 2009-01-15 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US9284409B2 (en) | 2007-07-19 | 2016-03-15 | Boston Scientific Scimed, Inc. | Endoprosthesis having a non-fouling surface |
| US7931683B2 (en) | 2007-07-27 | 2011-04-26 | Boston Scientific Scimed, Inc. | Articles having ceramic coated surfaces |
| US20090138077A1 (en) * | 2007-07-27 | 2009-05-28 | Boston Scientific Scimed, Inc. | Articles having ceramic coated surfaces |
| US20090029077A1 (en) * | 2007-07-27 | 2009-01-29 | Boston Scientific Scimed, Inc. | Drug eluting medical devices having porous layers |
| US8815273B2 (en) | 2007-07-27 | 2014-08-26 | Boston Scientific Scimed, Inc. | Drug eluting medical devices having porous layers |
| US20090035448A1 (en) * | 2007-07-31 | 2009-02-05 | Boston Scientific Scimed, Inc. | Medical device coating by laser cladding |
| US8221822B2 (en) | 2007-07-31 | 2012-07-17 | Boston Scientific Scimed, Inc. | Medical device coating by laser cladding |
| US20100137977A1 (en) * | 2007-08-03 | 2010-06-03 | Boston Scientific Scimed, Inc. | Coating for Medical Device Having Increased Surface Area |
| US8900292B2 (en) | 2007-08-03 | 2014-12-02 | Boston Scientific Scimed, Inc. | Coating for medical device having increased surface area |
| US20180092987A1 (en) * | 2007-08-14 | 2018-04-05 | The Regents Of The University Of California | Hollow silica nanospheres and methods of making same |
| US10328160B2 (en) | 2007-08-14 | 2019-06-25 | The Regents Of The University Of California | Hollow silica nanospheres and methods of making same |
| US20090104244A1 (en) * | 2007-09-21 | 2009-04-23 | Boston Scientific Scimed, Inc. | Therapeutic agent-eluting medical devices having textured polymeric surfaces |
| US7938855B2 (en) | 2007-11-02 | 2011-05-10 | Boston Scientific Scimed, Inc. | Deformable underlayer for stent |
| US20090118820A1 (en) * | 2007-11-02 | 2009-05-07 | Boston Scientific Scimed, Inc. | Deformable underlayer for stent |
| US8029554B2 (en) | 2007-11-02 | 2011-10-04 | Boston Scientific Scimed, Inc. | Stent with embedded material |
| US20090118822A1 (en) * | 2007-11-02 | 2009-05-07 | Holman Thomas J | Stent with embedded material |
| US20090118818A1 (en) * | 2007-11-02 | 2009-05-07 | Boston Scientific Scimed, Inc. | Endoprosthesis with coating |
| US20090118809A1 (en) * | 2007-11-02 | 2009-05-07 | Torsten Scheuermann | Endoprosthesis with porous reservoir and non-polymer diffusion layer |
| US8216632B2 (en) | 2007-11-02 | 2012-07-10 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US8920491B2 (en) | 2008-04-22 | 2014-12-30 | Boston Scientific Scimed, Inc. | Medical devices having a coating of inorganic material |
| US8932346B2 (en) | 2008-04-24 | 2015-01-13 | Boston Scientific Scimed, Inc. | Medical devices having inorganic particle layers |
| US8449603B2 (en) | 2008-06-18 | 2013-05-28 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US20090324684A1 (en) * | 2008-06-25 | 2009-12-31 | Boston Scientific Scimed, Inc. | Medical devices having surface coatings |
| US8277833B2 (en) | 2008-06-25 | 2012-10-02 | Boston Scientific Scimed, Inc. | Medical devices having surface coatings |
| US20090324685A1 (en) * | 2008-06-26 | 2009-12-31 | Boston Scientific Scimed, Inc. | Medical device coatings containing charged materials |
| US20100087783A1 (en) * | 2008-10-07 | 2010-04-08 | Boston Scientific Scimed, Inc. | Medical devices for delivery of therapeutic agents to body lumens |
| US20100137978A1 (en) * | 2008-12-03 | 2010-06-03 | Boston Scientific Scimed, Inc. | Medical Implants Including Iridium Oxide |
| US8231980B2 (en) | 2008-12-03 | 2012-07-31 | Boston Scientific Scimed, Inc. | Medical implants including iridium oxide |
| US20100228341A1 (en) * | 2009-03-04 | 2010-09-09 | Boston Scientific Scimed, Inc. | Endoprostheses |
| US8071156B2 (en) | 2009-03-04 | 2011-12-06 | Boston Scientific Scimed, Inc. | Endoprostheses |
| US20100274352A1 (en) * | 2009-04-24 | 2010-10-28 | Boston Scientific Scrimed, Inc. | Endoprosthesis with Selective Drug Coatings |
| US8287937B2 (en) | 2009-04-24 | 2012-10-16 | Boston Scientific Scimed, Inc. | Endoprosthese |
| US20100272882A1 (en) * | 2009-04-24 | 2010-10-28 | Boston Scientific Scimed, Inc. | Endoprosthese |
| US20140202759A1 (en) * | 2010-12-15 | 2014-07-24 | Advanced Bionics Ag | Protection for implanted gold surfaces |
| US9930819B2 (en) * | 2010-12-15 | 2018-03-27 | Advanced Bionics Ag | Protection for implanted gold surfaces |
| US9827337B2 (en) | 2011-06-22 | 2017-11-28 | J. Stefan Institute | Cathepsin-binding compounds bound to a carrier and their diagnostic use |
| WO2012175223A1 (en) * | 2011-06-22 | 2012-12-27 | J. Stefan Institute | Cathepsin-binding compounds bound to a carrier and their diagnostic use |
| US10668175B2 (en) | 2011-06-22 | 2020-06-02 | J. Stefan Institute | Cathepsin-binding compounds bound to a carrier and their diagnostic use |
| EP2537532A1 (en) * | 2011-06-22 | 2012-12-26 | J. Stefan Institute | Cathepsin-binding compounds bound to a nanodevice and their diagnostic and therapeutic use |
| US20140271901A1 (en) * | 2013-03-15 | 2014-09-18 | Mark Humayun | Management of Tractional Membranes |
| US11103593B2 (en) | 2013-10-15 | 2021-08-31 | Seagen Inc. | Pegylated drug-linkers for improved ligand-drug conjugate pharmacokinetics |
| US11919836B2 (en) | 2015-09-15 | 2024-03-05 | Praxis Bioresearch, LLC | Prodrugs of fencamfamine |
| US11229708B2 (en) | 2015-12-04 | 2022-01-25 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11793880B2 (en) | 2015-12-04 | 2023-10-24 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11844839B2 (en) | 2016-03-25 | 2023-12-19 | Seagen Inc. | Process for the preparation of pegylated drug-linkers and intermediates thereof |
| US11730822B2 (en) | 2017-03-24 | 2023-08-22 | Seagen Inc. | Process for the preparation of glucuronide drug-linkers and intermediates thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US6680047B2 (en) | 2004-01-20 |
| US6331289B1 (en) | 2001-12-18 |
| US20020102217A1 (en) | 2002-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6680047B2 (en) | Diagnostic/therapeutic agents | |
| US6264917B1 (en) | Targeted ultrasound contrast agents | |
| US6261537B1 (en) | Diagnostic/therapeutic agents having microbubbles coupled to one or more vectors | |
| US8454582B2 (en) | Methods and devices for the treatment of ocular conditions | |
| US20220047505A1 (en) | Controlled Absorption Water-Soluble Pharmaceutically Active Organic Compound Formulation for Once-Daily Administration | |
| US8158152B2 (en) | Lyophilization process and products obtained thereby | |
| US9016221B2 (en) | Surface topographies for non-toxic bioadhesion control | |
| DE69735901T2 (en) | ENHANCING OR IN CONNECTION WITH DIAGNOSTIC / THERAPEUTIC AGENTS | |
| US7905852B2 (en) | Skin-contacting-adhesive free dressing | |
| US7199151B2 (en) | DHA-pharmaceutical agent conjugates of taxanes | |
| US8314077B2 (en) | Fatty acid-pharmaceutical agent conjugates | |
| US20180015172A1 (en) | Engineered receptor/ligand system for delivery of therapeutic agents | |
| EP1054998A1 (en) | Transglutaminase linkage of agents to tissue | |
| EP1203141A1 (en) | Linkage of agents to tissue | |
| EP0991427A2 (en) | Improvements in or relating to diagnostic/therapeutic agents | |
| AU1632801A (en) | DHA-pharmaceutical agent conjugates | |
| World Health Organization | The use of common stems in the selection of International Nonproprietary names (INN) for pharmaceutical substances: 2002 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: GE HEALTHCARE AS,NORWAY Free format text: CHANGE OF NAME;ASSIGNORS:AMERSHAM HEALTH AS;NYCOMED IMAGING AS;REEL/FRAME:018039/0537 Effective date: 20060329 Owner name: GE HEALTHCARE AS, NORWAY Free format text: CHANGE OF NAME;ASSIGNORS:AMERSHAM HEALTH AS;NYCOMED IMAGING AS;REEL/FRAME:018039/0537 Effective date: 20060329 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |